Iperplasia Surrenalica Congenita
L’iperplasia surrenalica congenita (CAH), nota storicamente anche come sindrome adrenogenitale, è una famiglia di disordini autosomici recessivi caratterizzati da un difetto enzimatico nella via di biosintesi degli ormoni steroidei surrenalici. Nel 95% dei casi, la condizione è dovuta al deficit dell’enzima 21-idrossilasi.
Fisiopatologia
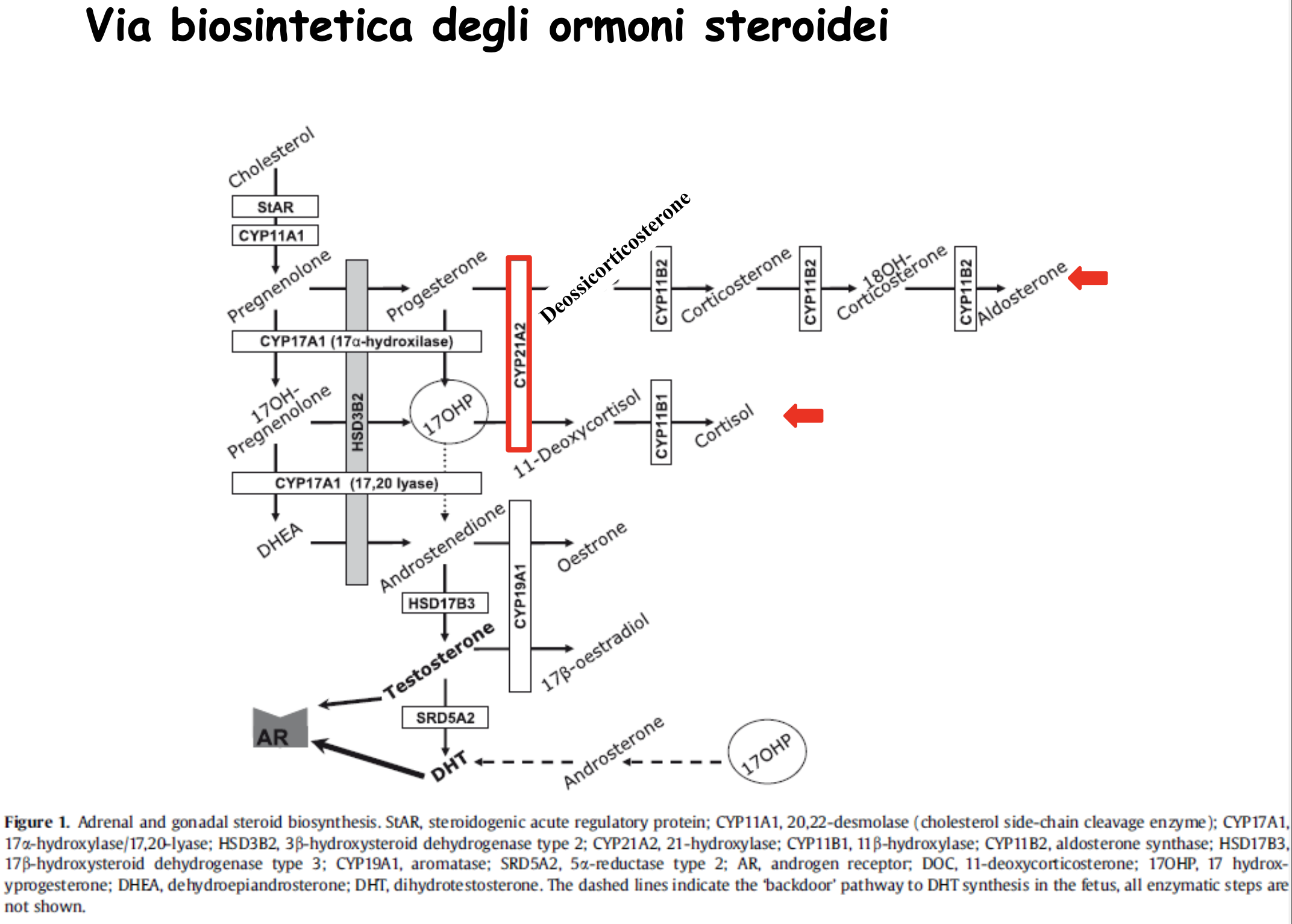
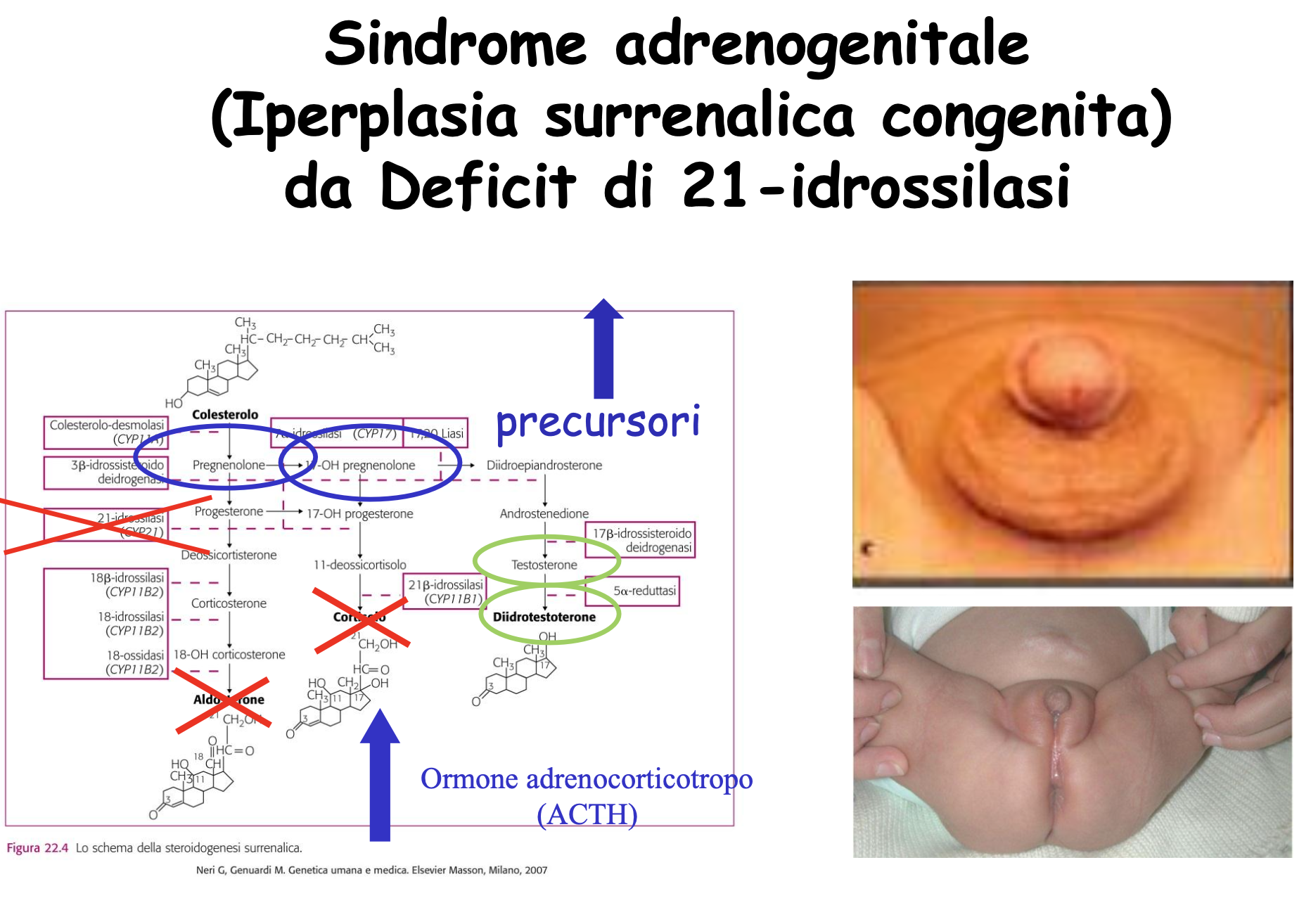
L’enzima 21-idrossilasi (codificato dal gene CYP21A2) converte il progesterone in deossicorticosterone (via dell’aldosterone) e il 17-idrossiprogesterone (17-OHP) in 11-deossicortisolo (via del cortisolo). Il blocco di questo enzima determina:
- Carenza di cortisolo: Rimuove il feedback negativo sull’ipofisi, che risponde secernendo massicce quantità di ormone adrenocorticotropo (ACTH).
- Iperplasia della corteccia surrenale: L’eccesso di ACTH stimola la crescita e l’attività iperplastica delle ghiandole surrenali.
- Accumulo di precursori steroidei (17-OHP): Non potendo progredire verso la sintesi di cortisolo e aldosterone, i precursori si accumulano e vengono deviati (shunt) in massa verso la sintesi di androgeni surrenalici (testosterone e DHT).
- Effetti clinici della virilizzazione: L’eccesso di androgeni surrenalici durante lo sviluppo fetale determina la virilizzazione (mascolinizzazione) dei genitali esterni nei feti femmine (
46,XX), portando a genitali ambigui alla nascita (ipertrofia clitoridea, fusione parziale o completa delle labbra). Nei feti maschi (46,XY) non si osservano anomalie dei genitali esterni alla nascita, ma pubertà precoce nell’infanzia. - Perdita di sali (forma classica grave): La carenza concomitante di aldosterone impedisce il riassorbimento renale di sodio, causando iponatriemia severa, iperpotassiemia, disidratazione e shock cardiocircolatorio nei primi giorni di vita (crisi surrenalica “salt-wasting”).
Genetica Molecolare del Locus 6p21
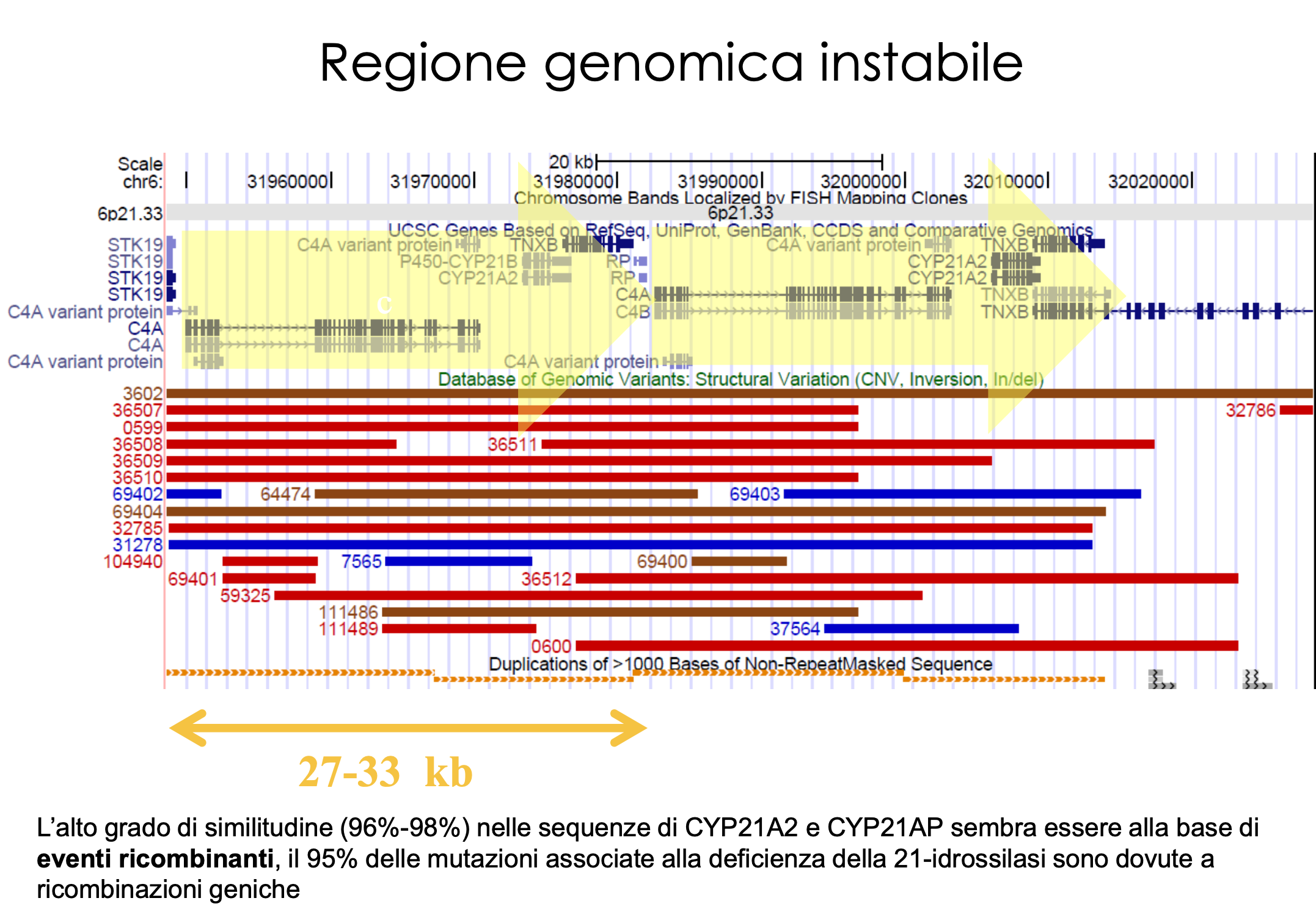
Il gene CYP21A2 è situato sul braccio corto del cromosoma 6 () all’interno del locus genico del complesso maggiore di istocompatibilità (HLA). È una delle regioni genomiche più complesse e instabili del genoma umano a causa della presenza di una duplicazione segmentale altamente omologa (modulo RCCX):
- Gene Funzionale: CYP21A2 (attivo).
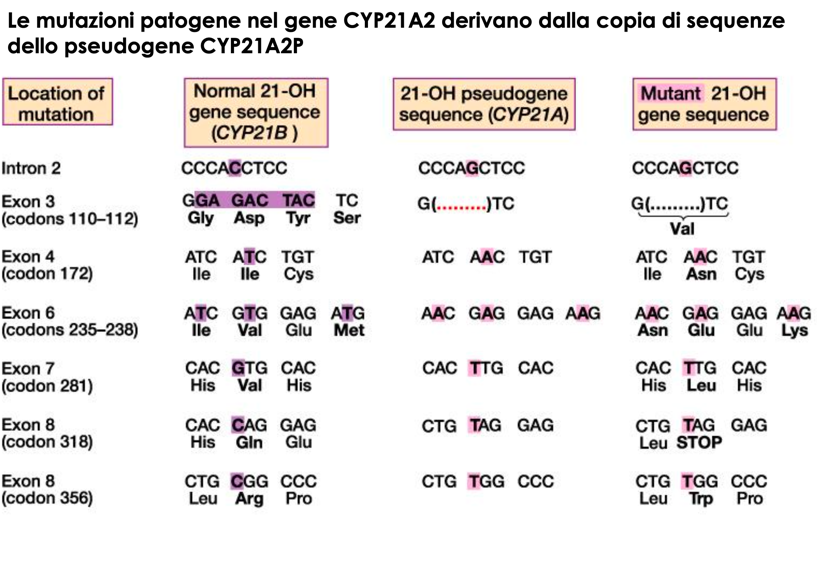
- Pseudogene: CYP21P (non funzionale). Presenta un’omologia di sequenza del 98% con il gene attivo ma contiene circa 10 mutazioni inattivanti (mutazioni deleterie, codoni di stop prematuri, delezioni frameshift).
- Geni Adiacenti: I geni duplicati C4A e C4B (componenti del complemento) e TNXB/TNXA.

Meccanismi Mutazionali Principali
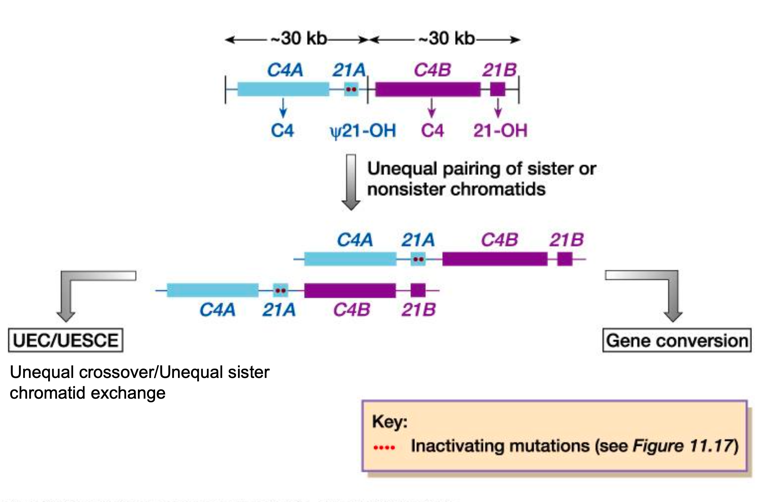
L’appaiamento errato meiotico tra il gene funzionale e lo pseudogene è favorito dalla stretta vicinanza e dall’elevata omologia:
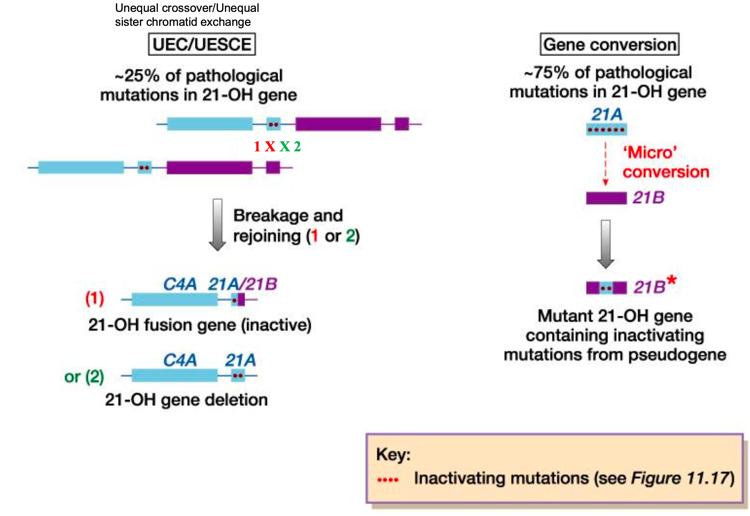
- Crossing Over Ineguale (25% dei casi): Genera gameti ricombinanti che portano o la delezione completa del gene CYP21A2 o la formazione di un gene chimerico inattivo CYP21P/CYP21A2 (contenente le mutazioni dello pseudogene).
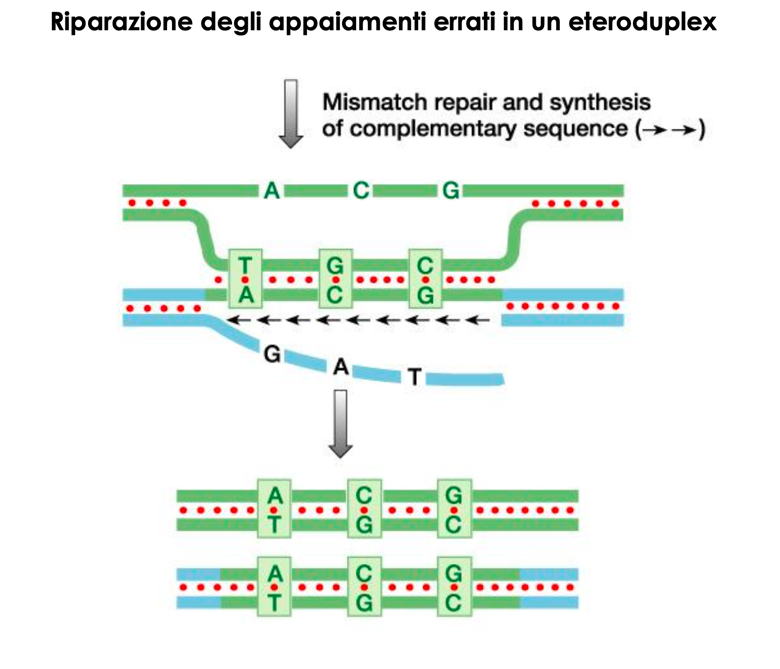
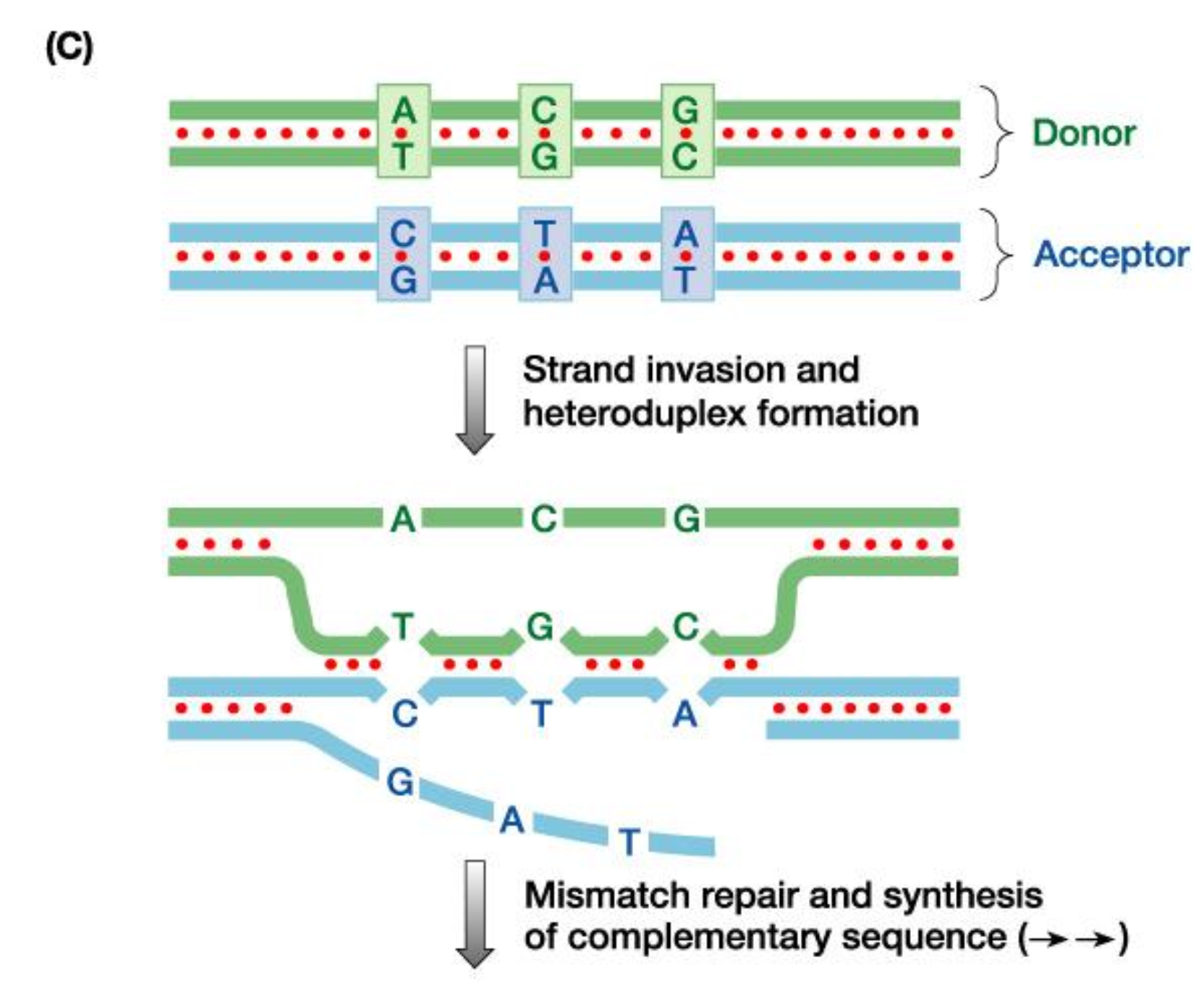
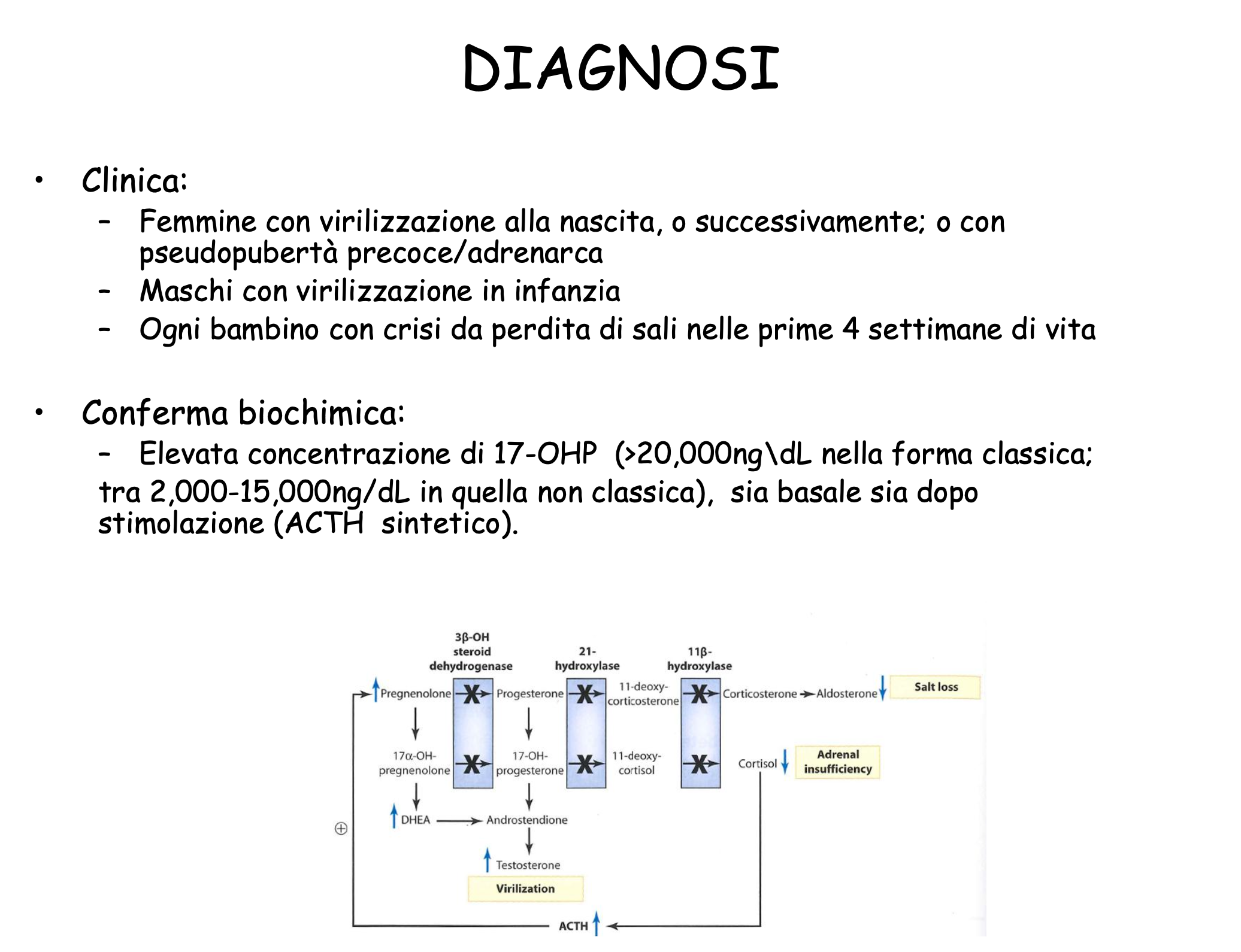
- Conversione Genica (75% dei casi): Trasferimento non reciproco e unidirezionale di sequenze dallo pseudogene CYP21P verso il gene attivo CYP21A2. Avviene per la formazione di un eteroduplex durante la meiosi, riparato dal sistema di mismatch repair utilizzando la sequenza dello pseudogene come stampo. Questo meccanismo fa sì che il 95% dei pazienti affetti presenti mutazioni tipiche dello pseudogene nel gene attivo CYP21A2.
Diagnosi
Diagnosi Molecolare
La diagnostica genetica prevede un approccio mirato:
- PCR target: Ricerca mirata delle mutazioni note tipiche dello pseudogene CYP21P trasferite per conversione genica.
- Analisi di delezione/dosaggio genico: Per identificare delezioni complete o geni chimerici.
- Sequenziamento esonico completo: Riservato al restante 5% dei casi che presentano varianti private non derivanti dallo pseudogene.
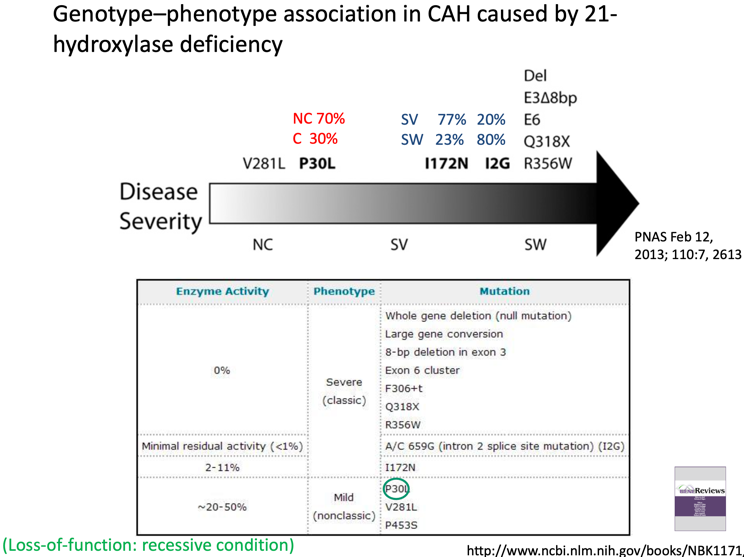
Diagnosi Biochimica
La conferma clinica viene effettuata dosando i livelli di 17-idrossiprogesterone (17-OHP) circolante:
- Valutazione effettuata sia a livello basale sia dopo stimolazione con ACTH sintetico. Consente di discriminare nettamente i pazienti con forma classica (valori elevatissimi), forma non classica (valori intermedi), soggetti portatori sani ed individui sani.
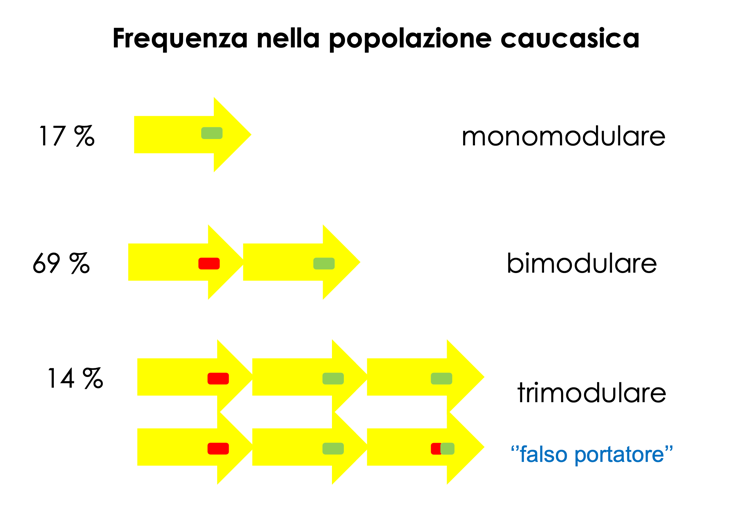
Variabilità Strutturale e Calcolo del Rischio Genetico
L’instabilità del locus genera aplotipi con diverso numero di moduli duplicati nella popolazione:
- Aplotipo Bimodulare (Classico): Presenza di uno pseudogene e un gene attivo in ciascun cromosoma.
- Aplotipo Monomodulare: Perdita dello pseudogene per delezione meiotica (fenotipicamente normale).
- Aplotipo Trimodulare: Presenza di tre moduli (es. due pseudogeni ed un gene attivo, oppure uno pseudogene e due geni attivi, uno dei quali mutato).
Rischio di Falsi Negativi in Diagnosi Prenatale
L’identificazione di mutazioni nei genitori deve sempre confermare che le varianti siano situate su alleli diversi (in trans). La presenza di aplotipi trimodulari può determinare errori interpretativi:
- Un genitore può presentare due mutazioni in cis sullo stesso cromosoma (su un aplotipo trimodulare) e possedere una terza copia del gene normale attiva sul medesimo allele. Il test genetico standard potrebbe identificarlo erroneamente come portatore semplice di due mutazioni (e quindi presunto affetto o portatore a rischio di trasmissione in trans), quando in realtà è un carrier sano che trasmette le mutazioni in blocco.


Terapia e Trattamento Prenatale
- Terapia Sostitutiva Post-natale: Somministrazione a vita di glucocorticoidi (idrocortisone/desametasone) per sopprimere l’asse ACTH e mineralocorticoidi (fludrocortisone) per la perdita di sali.
- Chirurgia Plastica Ricostruttiva: Interventi di clitoroplastica e vaginoplastica eseguiti in centri specializzati per correggere le anomalie dei genitali esterni nelle femmine.
- Trattamento Prenatale: Nelle coppie a rischio (entrambi portatori sani), è possibile somministrare desametasone alla madre a partire dalla 6ª settimana di gestazione (prima che avvenga la virilizzazione fetale). Il desametasone attraversa la barriera placentare e sopprime la secrezione di ACTH nel feto, prevenendo la virilizzazione dei feti femmina.
- Limiti e Trial Clinici: Questa terapia è controversa ed associata a significativi effetti collaterali materni (ipertensione, diabete gestazionale, aumento di peso) e fetali a lungo termine, rendendola oggetto di discussione etica e clinica.
🔗 Collegamenti
- Determinazione del Sesso Fenotipico — ⬆️ causa