Online Mendelian Inheritance in Man (OMIM)
L’Online Mendelian Inheritance in Man (OMIM) è un database internazionale online di geni umani e fenotipi genetici, fondato dal Dr. Victor A. McKusick. Costituisce il catalogo di riferimento per le Malattie Genetiche monogeniche (mendeliane).
Storia e Statistiche del Genoma
Dalla sua creazione ad oggi si è registrato un incremento massivo della scoperta dei geni responsabili di malattie genetiche. Tuttavia, esiste un divario significativo tra la conoscenza dei geni e la comprensione del loro impatto fenotipico:
- Geni totali descritti: Circa 17.000 geni catalogati.
- Associazione molecolare-fenotipica: Di questi 17.000 geni, solo per circa 7.000 conosciamo sia il fenotipo patologico sia la base molecolare (la mutazione specifica che causa la malattia). Conoscere il meccanismo molecolare è essenziale per lo sviluppo di terapie mirate e prevenzione neonatale (es. limitazione dietetica di un substrato in una malattia metabolica).
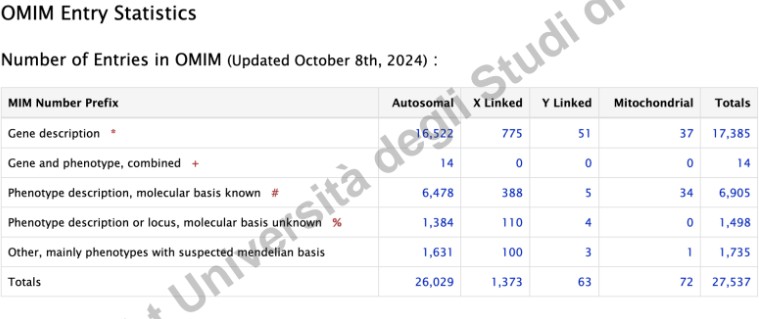
Distribuzione Funzionale dei Geni in Relazione alle Patologie
I dati mostrano la seguente ripartizione dell’impatto delle mutazioni geniche:
- 15%: Geni identificati come responsabili di malattie mendeliane note.
- 3%: Locus genico mappato (associato alla malattia), ma la mutazione causativa specifica non è ancora stata identificata.
- 30%: Geni la cui mutazione determina letalità embrionale (studiati prevalentemente tramite modelli animali come il topo). La letalità precoce impedisce l’associazione clinica a una malattia post-natale nell’uomo.
- 52%: Geni con impatto fenotipico sconosciuto (non è noto se una loro mutazione provochi o meno una patologia).
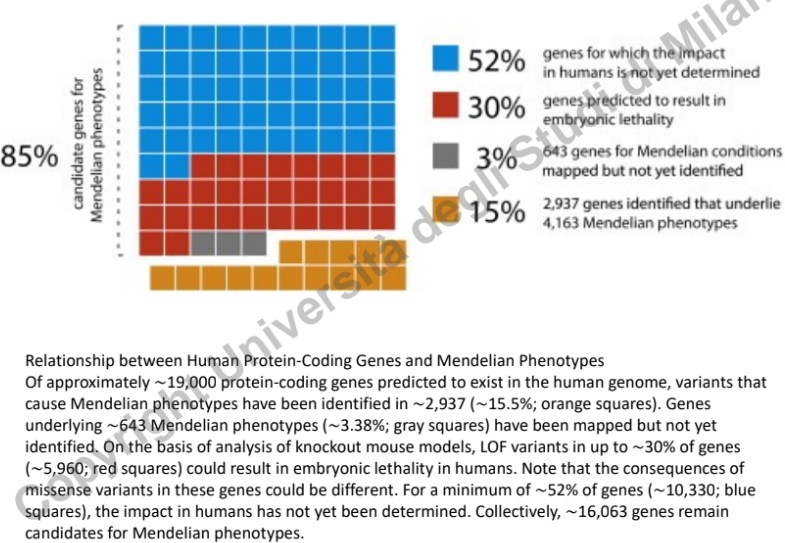
Tecnologie di Sequenziamento e Flusso di Validazione
L’avvento delle tecniche di sequenziamento di nuova generazione (NGS - Next Generation Sequencing), in particolare il Whole Genome Sequencing (WGS) e il Whole Exome Sequencing (WES), ha accelerato la scoperta di geni implicati in fenotipi mendeliani.
Il Processo di Validazione delle Varianti
Il sequenziamento dell’intero esoma o genoma di un individuo affetto produce migliaia di varianti genetiche. Identificare la singola mutazione causativa richiede un flusso di lavoro strutturato:
- Analisi In Silico: Utilizzo di algoritmi predittivi e simulazioni matematiche al computer per stimare la gravità della mutazione e classificarla come benigna o intollerata (patogenetica).
- Validazione In Vivo: Test funzionali su organismi modello (es. knock-out nel topo) per confermare il nesso causale tra gene e fenotipo.
- Limite clinico: La validazione in vivo è particolarmente complessa per patologie a forte componente comportamentale o psichiatrica (es. schizofrenia, autismo), difficilmente riproducibili negli organismi modello.
🔗 Collegamenti
- Malattie Genetiche — ⬆️ causa
- Progetto Genoma Umano — 🔗 stesso meccanismo / stessa via