Sindrome da Tremore e Atassia associata a X Fragile
La sindrome da tremore e atassia associata a X fragile (FXTAS, Fragile X-associated Tremor/Ataxia Syndrome) è un disturbo neurodegenerativo tardivo correlato allo stato di premutazione del gene FMR1. Colpisce prevalentemente i soggetti di sesso maschile in età matura (generalmente dopo i 50-60 anni).
Quadro Clinico ed Epidemiologia
La sintomatologia clinica è progressiva e si caratterizza per:
- Tremore intenzionale (evidente durante l’esecuzione di movimenti mirati).
- Atassia cerebellare e gravi problemi di bilanciamento/coordinazione motoria.
- Deficit cognitivi, ad esordio sfumato, che possono progredire fino a demenza terminale.
Epidemiologia e Penetranza legata all’Età:
La penetranza della FXTAS aumenta con l’età nei soggetti portatori della premutazione:
- All’età di 50 anni, circa il 30% dei soggetti maschi con premutazione presenta manifestazioni cliniche. La frequenza aumenta progressivamente nei decenni successivi.
- Le donne portatrici di premutazione mostrano una frequenza e una severità clinica nettamente inferiori rispetto ai maschi. Questo è dovuto all’Inattivazione del Cromosoma X (mosaicismo cellulare), in cui la presenza del cromosoma X sano vicaria in parte il deficit del cromosoma mutato.
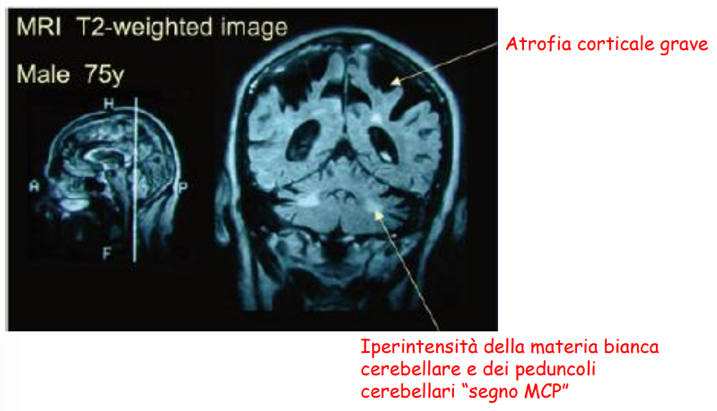
Diagnosi strumentale (Neuroimaging)
La diagnosi precoce della FXTAS può essere supportata da esami di risonanza magnetica cerebrale (RMN), che evidenziano reperti caratteristici correlati alla progressione del danno:
- Fase Precoce: Presenza di iperintensità bilaterale della sostanza bianca cerebellare e dei peduncoli cerebellari medi (MCP sign).
- Fase Avanzata: Sviluppo di una diffusa e grave atrofia corticale.
Patogenesi Molecolare: Tossicità da RNA e Feedback Trascrizionale
A differenza della Sindrome dell’X Fragile (dovuta a perdita di funzione per silenziamento epigenetico del gene con triplette), la FXTAS è causata da un meccanismo di tossicità dell’mRNA:
- Mancato Silenziamento: Nello stato di premutazione (triplette CGG comprese solitamente tra 80 e 100 unità nella Sbobina 28, o più in generale 55-199), l’espansione non è tale da indurre la chiusura della cromatina; il gene FMR1 viene quindi attivamente trascritto.
- Difficoltà di Traduzione: La presenza delle triplette espanse CGG nella porzione 5’ UTR dell’mRNA rende la traduzione ribosomiale della proteina FMRP estremamente difficoltosa e inefficiente.
- Feedback Positivo Trascrizionale: La ridotta quantità di proteina FMRP prodotta stimola un meccanismo di feedback positivo a livello nucleare per compensare il deficit, inducendo un forte aumento della trascrizione del gene FMR1.
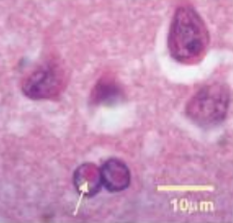
- Accumulo di mRNA e Inclusioni Eosinofile: L’eccesso di mRNA mutato trascritto non riesce ad essere tradotto e forma complessi insolubili che precipitano all’interno del nucleo e del citoplasma di neuroni e astrociti. Istologicamente, questo accumulo si apprezza sotto forma di inclusioni eosinofile patognomoniche, le quali inducono sofferenza e successiva morte cellulare nei tessuti cerebrali.
Caso Familiare
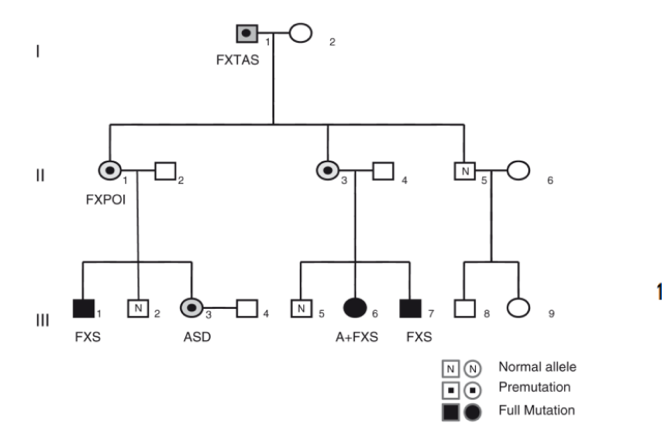
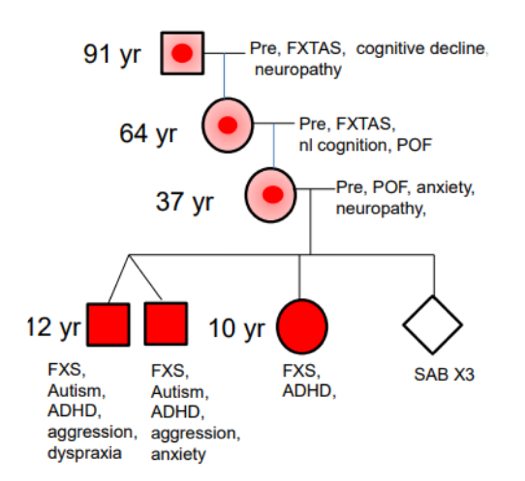
Nei pedigree familiari, si osserva frequentemente la co-segregazione di soggetti anziani con FXTAS (es. bisnonni portatori di premutazione) e nipoti o pronipoti affetti da sindrome dell’X fragile (full mutation) o disturbi correlati (es. autismo, ADHD, ASD), a causa dell’espansione meiotica femminile.
🔗 Collegamenti
- Sindrome dell’X Fragile — 📋 fa parte di
- Mutazioni Dinamiche — 📋 fa parte di
- Insufficienza Ovarica Prematura associata a X Fragile — 🔄 diagnosi differenziale / confronto
- Inattivazione del Cromosoma X — 🔄 diagnosi differenziale / confronto