Proteina MeCP2
La proteina MeCP2 (da Methyl-CpG-binding protein 2) è il principale lettore (reader) epigenetico del DNA metilato. È un fattore di trascrizione ad attività repressiva ubiquitario, essenziale per la maturazione del sistema nervoso centrale (SNC) e per la regolazione fine dell’espressione genica nei neuroni.
Struttura della Proteina
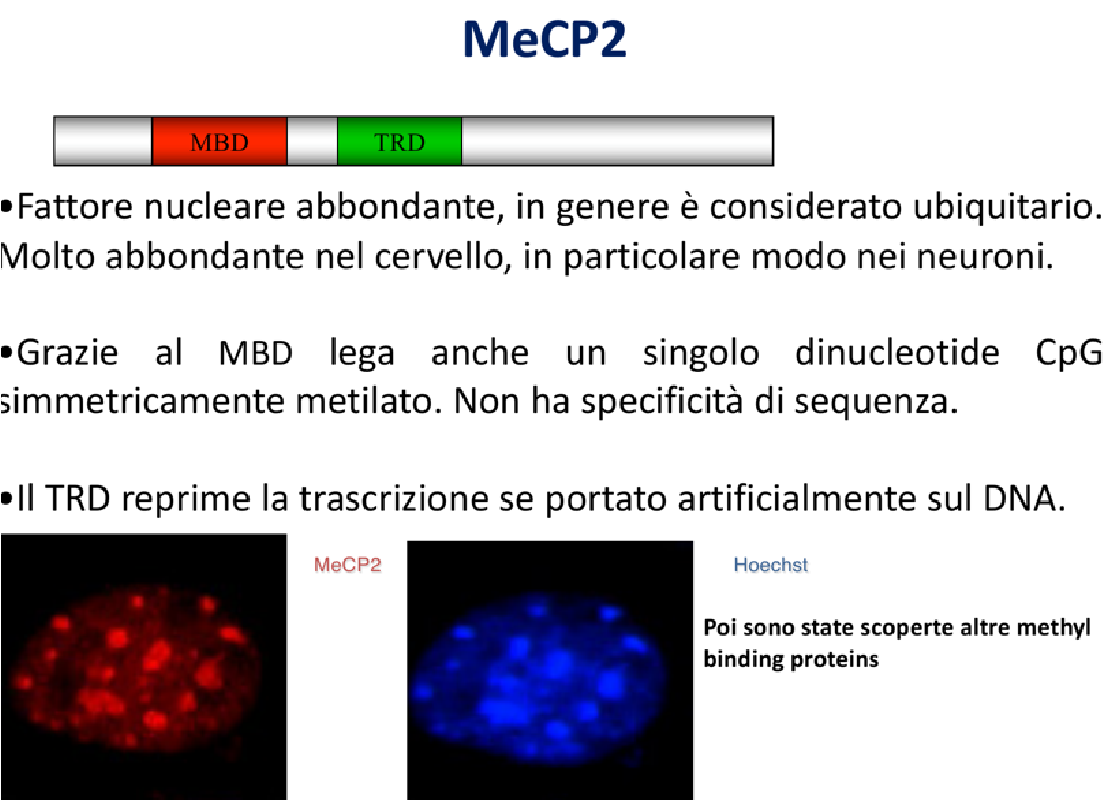
MeCP2 è una proteina di circa 485-500 amminoacidi con una struttura modulare caratterizzata da due domini funzionali principali:
- MBD (Methyl-CpG-Binding Domain): Dominio globulare in grado di riconoscere e legare specificamente il DNA metilato (e non una specifica sequenza nucleotidica).
- TRD (Transcriptional Repression Domain): Dominio deputato alla repressione trascrizionale attiva. Una volta che la proteina è legata al DNA, il TRD recluta enzimi deacetilasi istonici (HDAC) o complessi di rimodellamento cromatinico per indurre lo stato eterocromatico chiuso.
I domini sono autosufficienti e mantengono la loro funzionalità anche se rimossi o fusi ad altre proteine.
Abbondanza e Funzione Fisiologica nel Cervello
MeCP2 è considerata una proteina ubiquitaria, ma risulta espressa in quantità estremamente elevate a livello dei neuroni cerebrali:
- Sostituzione dell’Istone H1: Nei neuroni maturi, la concentrazione di MeCP2 è così elevata che la proteina si sostituisce all’istone linker H1.
- Organizzazione della Cromatina: Questa elevatissima densità determina una struttura di cromatina neuronale peculiare e differente da quella delle altre cellule somatiche, fondamentale per garantire un’espressione genica estremamente controllata in risposta all’attività sinaptica.
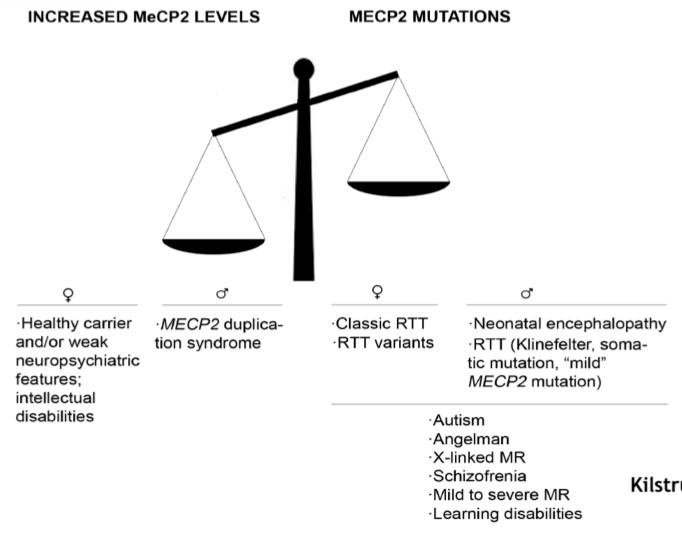
Sensibilità al Dosaggio Genico
La funzionalità del cervello dipende da una stretta regolazione della quantità di MeCP2. Lo spettro di dosaggio della proteina è altamente sensibile:
- Loss-of-Function (Deficit): L’assenza o la ridotta attività di MeCP2 determina la Sindrome di Rett nelle femmine (eterozigoti) e quadri gravi di encefalopatia neonatale nei maschi.
- Gain-of-Function (Overdose): Un eccesso della proteina è altrettanto tossico. L’overdose genica determina la sindrome da duplicazione di MeCP2 (caratterizzata da grave disabilità intellettuale, epilessia e infezioni ricorrenti nei maschi). Sperimentalmente, topi sani a cui viene data una copia extra di MeCP2 performano inizialmente meglio nei test cognitivi, per poi andare incontro a un rapido declino neurologico e alla morte.
Mutazioni Patologiche
La gravità clinica dipende dal tipo di mutazione del gene MECP2 (localizzato sul cromosoma X):
- Hot-spot Mutazionali: Molte mutazioni missenso colpiscono il dominio MBD, impedendo alla proteina di legare il DNA metilato.
- Mutazioni C-terminali: Anche piccole delezioni a livello dell’estremità C-terminale (come la perdita degli ultimi 4 amminoacidi) sono sufficienti a inattivare la proteina e a determinare lo sviluppo della sindrome di Rett.
Un individuo su 6000 a livello mondiale presenta patologie neuropsichiatriche o del neurosviluppo associate ad alterazioni del gene MECP2.
🔗 Collegamenti
- Proteine MBD — 📋 fa parte di
- Sindrome di Rett — ⬇️ conseguenza
- Metilazione del DNA — 🔗 stesso meccanismo
- Istone H1 — 🔄 diagnosi differenziale / confronto