Sindrome di Rett
La sindrome di Rett è una grave malattia genetica del neurosviluppo che colpisce prevalentemente il sesso femminile (incidenza di circa 1 su 10.000 bambine). Rappresenta la prima causa di grave disabilità intellettuale nelle femmine.
Eziologia e Genetica
La patologia è causata da mutazioni a carico del gene MECP2 (localizzato sul cromosoma X), che codifica per la Proteina MeCP2, un lettore essenziale della metilazione del DNA.
- Mutazioni Sporadiche: Nella stragrande maggioranza dei casi (oltre il 95%), la mutazione è di tipo sporadico e insorge de novo a livello dei gameti maschili (nello sperma).
- Espressione Eterozigote (Femmine): Le pazienti femmine sono eterozigoti per la mutazione. Per via dell’inattivazione casuale del cromosoma X (mosaicismo cellulare), coesistono cellule che esprimono l’allele sano e cellule che esprimono l’allele mutato, il che consente la sopravvivenza.
- Espressione Emizigote (Maschi): Poiché i maschi possiedono un solo cromosoma X, le mutazioni loss-of-function in MECP2 determinano un quadro gravissimo di encefalopatia neonatale, con ipotonia, epilessia severa e decesso precoce (generalmente entro i 10 anni). I maschi possono manifestare la sindrome di Rett classica solo in casi particolari:
- Presenza della sindrome di Klinefelter (corredo cromosomico XXY, in cui uno dei due cromosomi X porta l’allele sano).
- Presenza di mutazioni ipomorfe (poco gravi) o mosaicismi somatici.
Quadro Clinico
La malattia è caratterizzata da un decorso clinico tipico suddiviso in fasi:
- Sviluppo Apparentemente Normale (primi 6-36 mesi): Le bambine nascono senza difetti macroscopici e si sviluppano in maniera apparentemente sana. Tuttavia, un’analisi fine evidenzia anomalie precoci (es. vocalizzazioni a frequenze insolite o lievi alterazioni posturali inframezzate a comportamenti normali).
- Fase di Regressione Drammatica (criterio diagnostico obbligatorio): In un arco temporale ristretto (circa un mese), la bambina perde le capacità precedentemente acquisite (linguaggio verbale, disegno, motricità fine e coordinazione). Si riscontra perdita di interesse per l’ambiente circostante e forte irritabilità.
- Stereotipie delle Mani (criterio diagnostico obbligatorio): Insorgono movimenti compulsivi e ritmici involontari delle mani (es. battere le mani, stringerle o mordersele) che impediscono l’uso funzionale degli arti superiori.
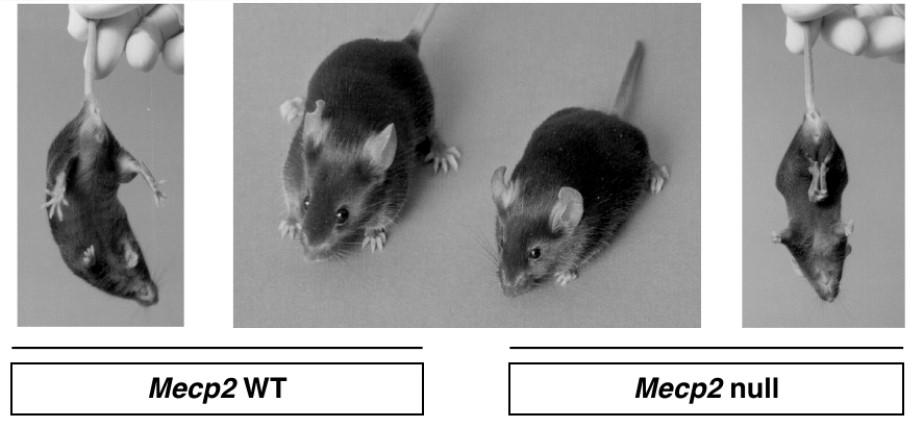
- Stabilizzazione e Sintomi Conclamati: La malattia si stabilizza, ma subentrano gravi complicanze tra cui disturbi respiratori (apnee e iperventilazione), crisi epilettiche farmaco-resistenti, grave scoliosi (con rotazioni della colonna fino a 100 gradi) e atrofia muscolare neurogena (dovuta a mancanza di impulsi corretti dal SNC, non a un difetto muscolare intrinseco). L’aspettativa di vita media si attesta intorno ai 50 anni, con totale dipendenza dai caregiver.

Morfologia Neuronale
Morfologicamente, il cervello delle pazienti è più piccolo (microcefalia acquisita), ma non si riscontra morte cellulare né gliosi (infiammazione). Le caratteristiche salienti dei neuroni includono:
- Soma (corpo cellulare) di dimensioni ridotte.
- Drastica riduzione delle ramificazioni dendritiche e delle spine sinaptiche.
- Errata espressione e repressione genica a livello neuronale in risposta agli stimoli ambientali.
Fattori che Influenzano la Gravità
La severità dei sintomi varia notevolmente a seconda di:
- Tipo di Mutazione: Sono descritte oltre 600 mutazioni in MECP2. Le mutazioni missenso cadono spesso nel dominio MBD. Anche piccolissime mutazioni, come la delezione degli ultimi 4 amminoacidi della proteina, causano la malattia.
- Inattivazione del Cromosoma X: Se l’inattivazione del cromosoma X è sbilanciata a favore dell’allele sano (inattivazione preferenziale del cromosoma X mutato), la paziente può mostrare sintomi lievi o essere una portatrice sana.
- Geni Modificatori: Varianti alleliche di altri geni che possono modulare (attenuare o esacerbare) il fenotipo clinico.
Diagnosi Molecolare
Consiste in:
- Prelievo di sangue venoso.
- Amplificazione degli esoni e delle giunzioni esone-introne del gene MECP2 tramite PCR.
- Sequenziamento per identificare mutazioni note in letteratura. In caso di mutazioni sconosciute, si sequenziano i genitori per stabilire se la mutazione è de novo (patogenica) o ereditata da un genitore sano (presunta variante non patogenica).
Reversibilità ed Evidenze Sperimentali
Per molti anni la sindrome di Rett è stata considerata incurabile. Nel 2007, un esperimento di Adrian Bird su modelli murini condizionali ha dimostrato che la patologia è reversibile:
- Il Modello Murino: Topi knockout per MeCP2 mostrano i medesimi sintomi dell’uomo (regressione a 5-6 settimane nel maschio, atassia, aprassia, stereotipie, disturbi respiratori e morte a 80 giorni).
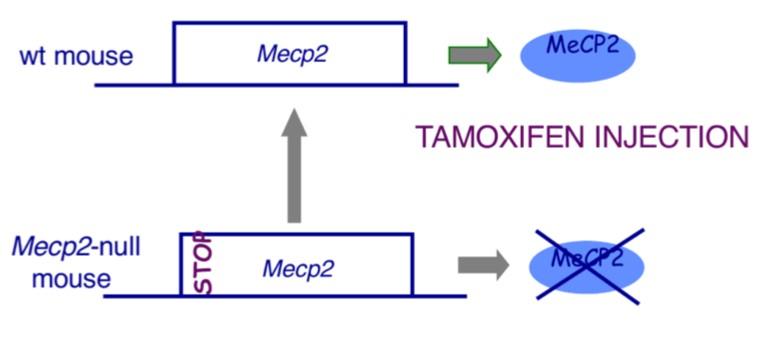
- Riattivazione Condizionale: Inserendo un gene MECP2 silenziato da un codone di stop che può essere rimosso farmacologicamente, Bird ha riattivato il gene in topi adulti già sintomatici.
- Risultati: Se la proteina viene riaccesa troppo velocemente, i topi muoiono a causa di cortocircuiti molecolari tra la proteina riattivata e i meccanismi compensatori attivati dalla cellula. Se invece la riattivazione avviene in modo graduale, i sintomi neurologici e respiratori regrediscono completamente e i topi riprendono a camminare normalmente.
- Significato Biologico: La sindrome di Rett è una patologia del neurosviluppo e non una malattia neurodegenerativa; i neuroni sono strutturalmente integri sebbene morfologicamente immaturi e possono essere ripristinati alla normale funzionalità.
Approcci Terapeutici
La ricerca clinica segue tre linee di sviluppo:
- Terapia Genica: Volta a reinserire il gene MECP2 funzionante. Richiede estrema cautela poiché lievi fluttuazioni o un eccesso di MeCP2 (overdose) sono altamente tossici per il cervello e per il fegato.
- Approcci Farmacologici: Diretti a modulare le vie di segnalazione alterate a valle di MeCP2.
- Approcci Molecolari:
- Trofinetide (Daybue): Il primo farmaco approvato (in USA) per il trattamento della sindrome di Rett. Non cura la patologia alla radice, ma apporta miglioramenti significativi nelle capacità motorie, comunicative e sociali.

🔗 Collegamenti
- Proteina MeCP2 — ⬆️ causa
- Metilazione del DNA — ⬆️ causa
- Mutazioni — 📋 fa parte di
- Sindrome ICF — 🔄 diagnosi differenziale / confronto