Ereditarietà Mitocondriale
L’ereditarietà mitocondriale (o matrilineare) è una modalità di trasmissione non mendeliana di caratteri o patologie causati da mutazioni a carico del DNA mitocondriale (mtDNA). Poiché i mitocondri dello zigote derivano quasi esclusivamente dall’ovocita, la trasmissione avviene unicamente per via materna.
Biologia del Genoma Mitocondriale (mtDNA)
Il mitocondrio è la centrale energetica cellulare. Contiene al suo interno da 5 a 10 molecole di DNA.
- Distribuzione cellulare: Il numero di mitocondri varia in base all’attività metabolica del tessuto. Ad esempio, i linfociti ne hanno pochi, le cellule muscolari ne contengono molti, mentre l’ovocita ne possiede oltre 100.000 per sostenere le prime divisioni embrionali. Globuli rossi e alcune cellule cheratinizzate della cute ne sono privi.
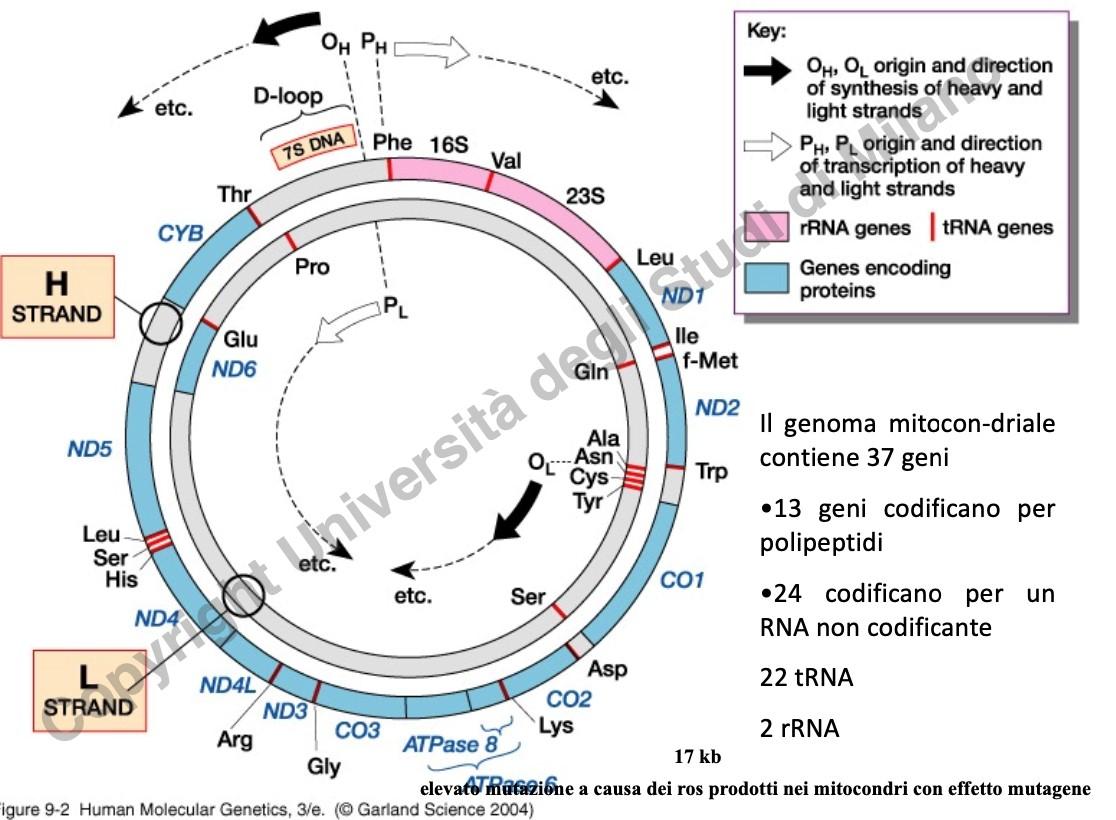
- Struttura molecolare: Il mtDNA è una molecola circolare a doppio filamento di circa 16.569 paia di basi (bp), formata da:
- Strand H (Heavy): Filamento pesante, ricco in guanine e citosine.
- Strand L (Light): Filamento leggero.
- Densità genica: È estremamente elevata (1 gene ogni 450 bp, a fronte di 1 gene ogni 40.000 bp del DNA nucleare). Ospita 37 geni:
- 24 geni per RNA non codificanti: 22 tRNA e 2 rRNA, necessari per la traduzione interna al mitocondrio.
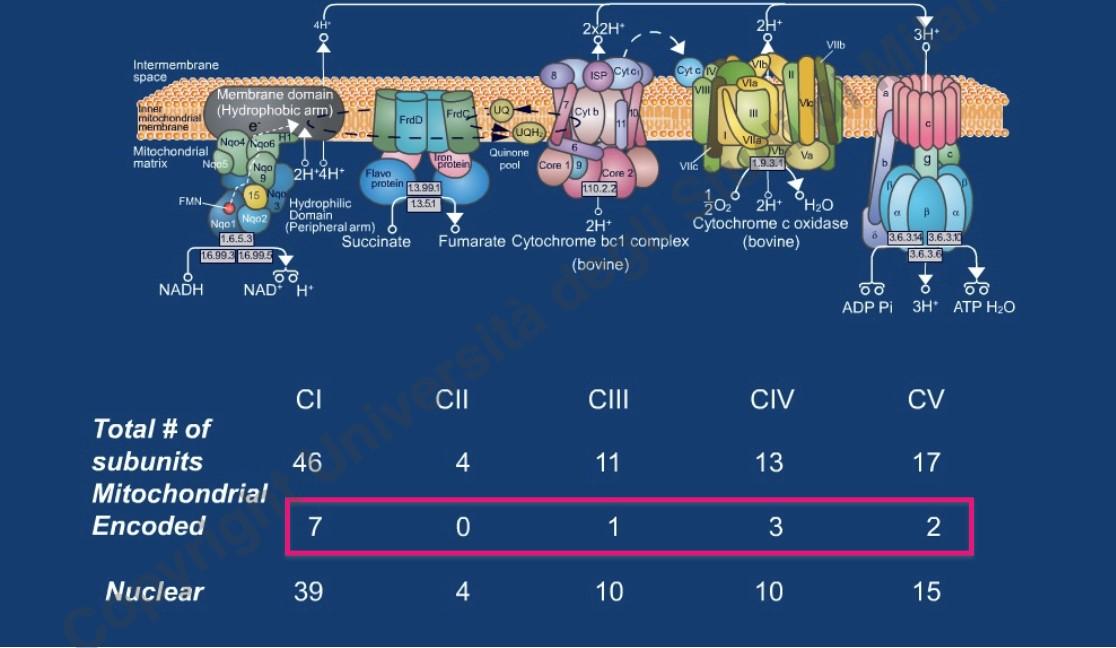
- 13 geni per polipeptidi: Subunità proteiche idrofobiche dei complessi della catena respiratoria (fosforilazione ossidativa).
Peculiarità Strutturali del mtDNA
- Assenza di introni.
- Parziale sovrapposizione tra le sequenze geniche.
- Trascrizione in forma di RNA policistronico.
- Mancanza di istoni (“DNA nudo”).
Suscettibilità alle Mutazioni
Il tasso di mutazione del mtDNA è significativamente più elevato rispetto a quello del DNA nucleare per tre motivi principali:
- Esposizione ai ROS: Il mtDNA è localizzato nella matrice mitocondriale, a diretto contatto con i radicali liberi dell’ossigeno generati dalla catena di trasporto degli elettroni.
- Assenza di istoni: Mancanza di una protezione fisica cromatinica.
- Riparazione inefficiente: Mancanza dei complessi sistemi di riparazione del DNA presenti nel nucleo.
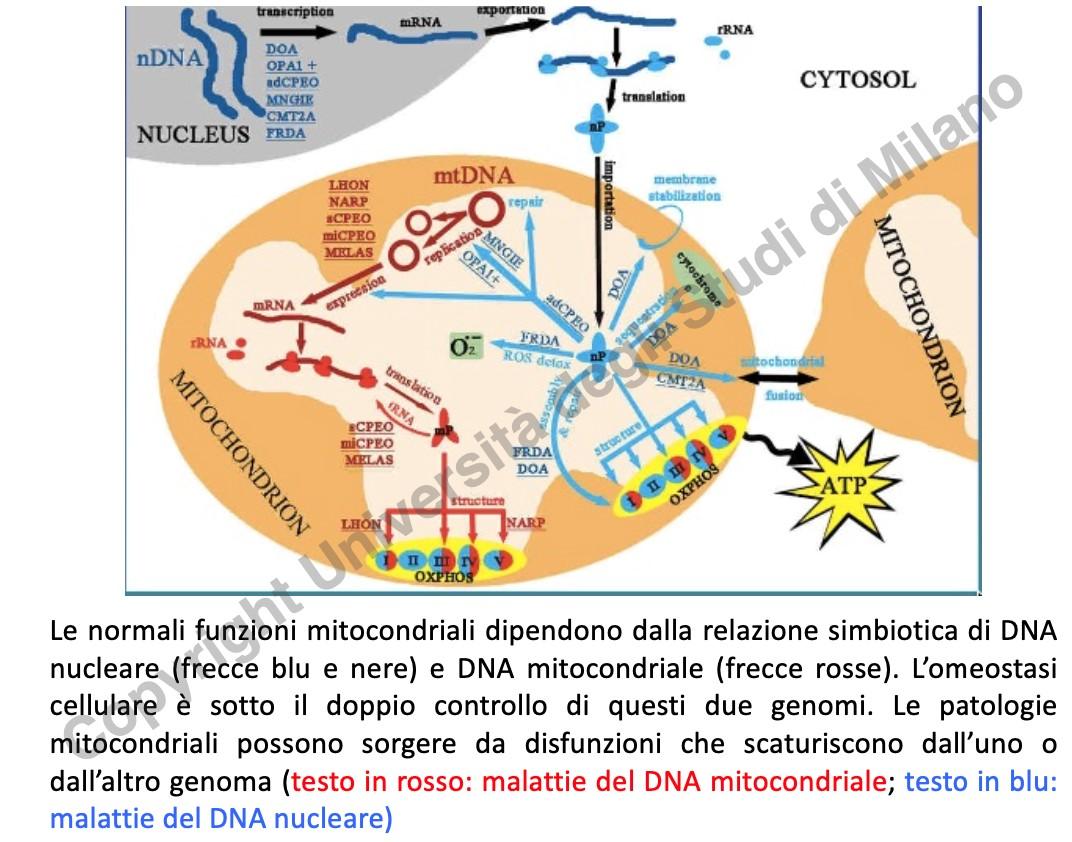
Genoma Nucleare vs Mitocondriale
La catena respiratoria è composta da circa 100 proteine. Solo 13 sono codificate dal mtDNA; le restanti 87 sono codificate dal DNA nucleare, sintetizzate nel citosol e importate nel mitocondrio. Le patologie mitocondriali possono quindi derivare sia da mutazioni del mtDNA sia da mutazioni del DNA nucleare (es. l’atassia di Friedreich o patologie congenite rare).
- Se la mutazione è nucleare, tutti i mitocondri cellulari saranno alterati.
- Se la mutazione è nel mtDNA, l’effetto dipenderà dalla percentuale di genomi mutati (eteroplasmia).
Caratteristiche dell’Ereditarietà del mtDNA
La genetica mitocondriale si basa su cinque regole fondamentali:
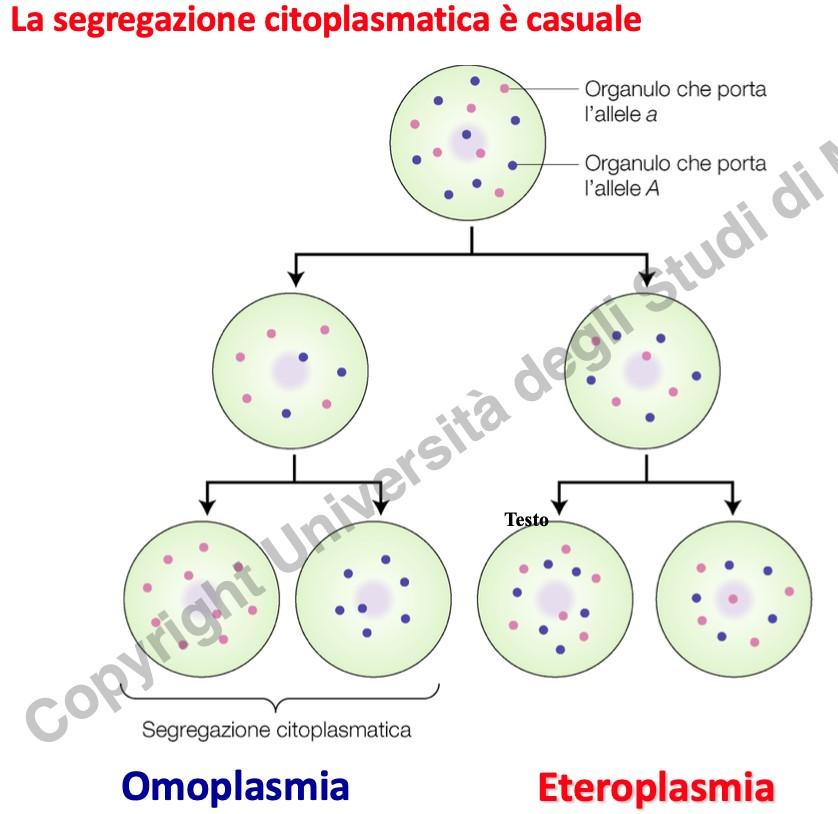
- Poliplasmia: Presenza di centinaia o migliaia di copie di mtDNA in ogni cellula.
- Segregazione Mitotica: Durante la citocinesi, la ripartizione dei mitocondri e delle rispettive molecole di mtDNA tra le cellule figlie avviene in modo casuale e asimmetrico, senza l’ausilio di un fuso mitotico. Ciò determina percentuali diverse di mtDNA mutato nelle cellule figlie.
- Eteroplasmia: Coesistenza, all’interno della stessa cellula o dello stesso mitocondrio, di molecole di mtDNA mutato e wild-type. L’omoplasmia (100% di mtDNA mutato o sano) è estremamente rara per mutazioni patologiche.
- Effetto Soglia: La disfunzione cellulare e la sintomatologia clinica si manifestano solo quando la percentuale di mtDNA mutato supera un limite critico (soglia), solitamente stimato sopra il 60-80%, al di sotto del quale la cellula compensa con il mtDNA sano.
- Trasmissione Matrilineare: Il fenotipo è trasmesso unicamente dalla madre. Gli spermatozoi contengono pochi mitocondri posizionati nel collo (funzionali alla motilità della coda) che vengono esclusi o attivamente degradati all’atto della fecondazione (in cui penetra solo la testa spermatica). Pertanto:
- Le femmine affette trasmettono la mutazione a tutti i figli (maschi e femmine).
- I maschi affetti non trasmettono la malattia a nessun discendente.
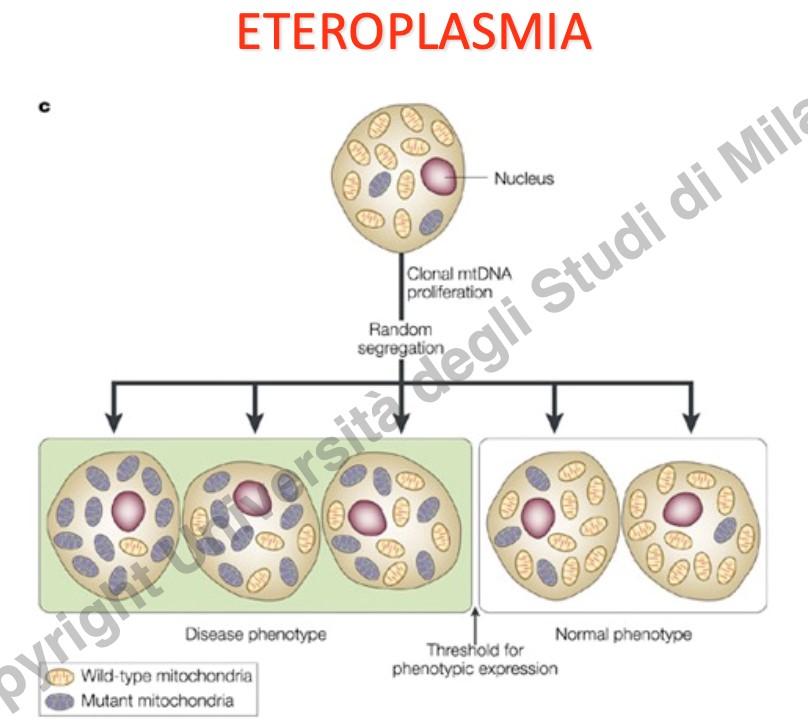
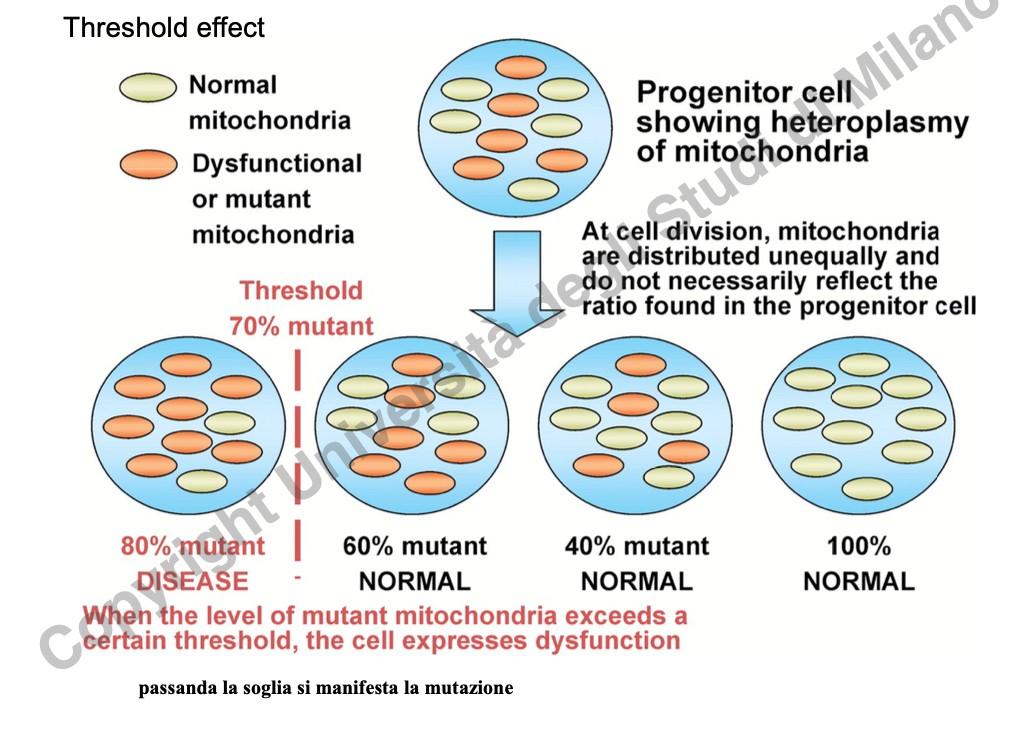
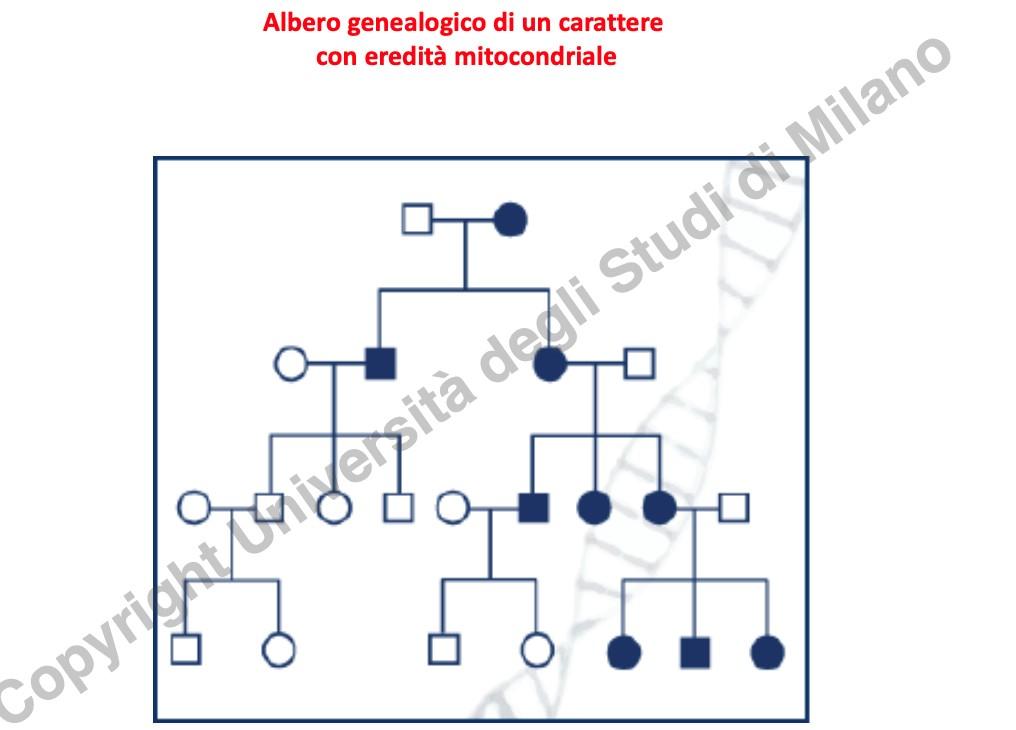
Eterogeneità Fenotipica e Collo di Bottiglia (Bottleneck)
Le malattie mitocondriali presentano un’estrema variabilità clinica intrafamiliare (es. nella sindrome di MERRF, dove membri della stessa famiglia manifestano sintomi che variano da sordità isolata a epilessia severa e demenza).
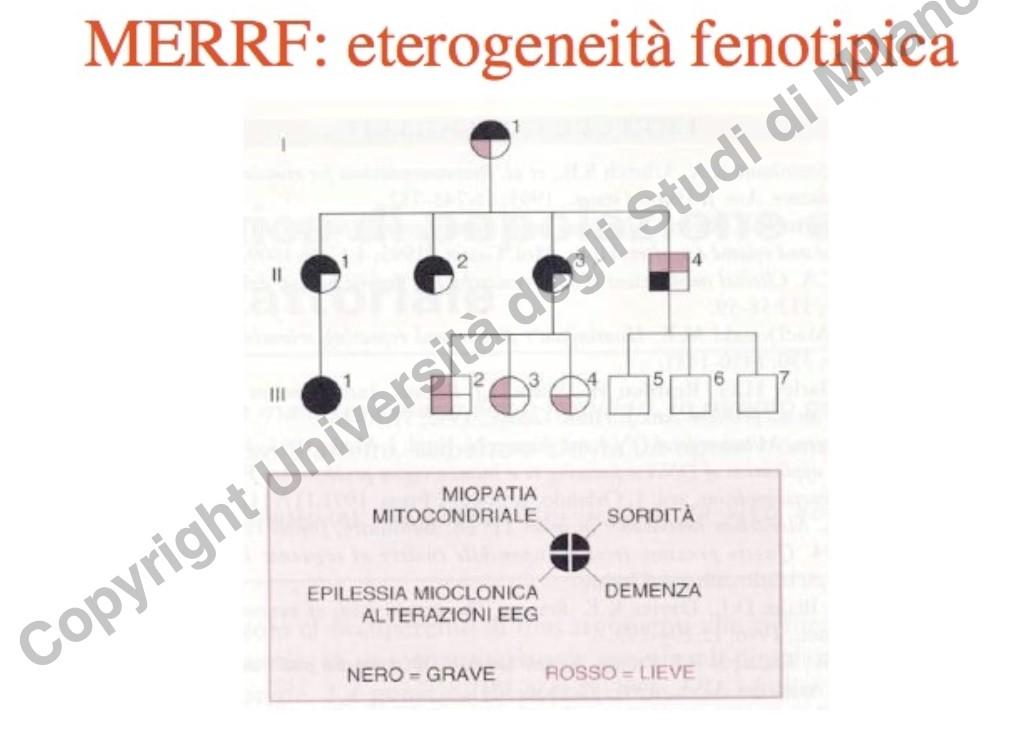
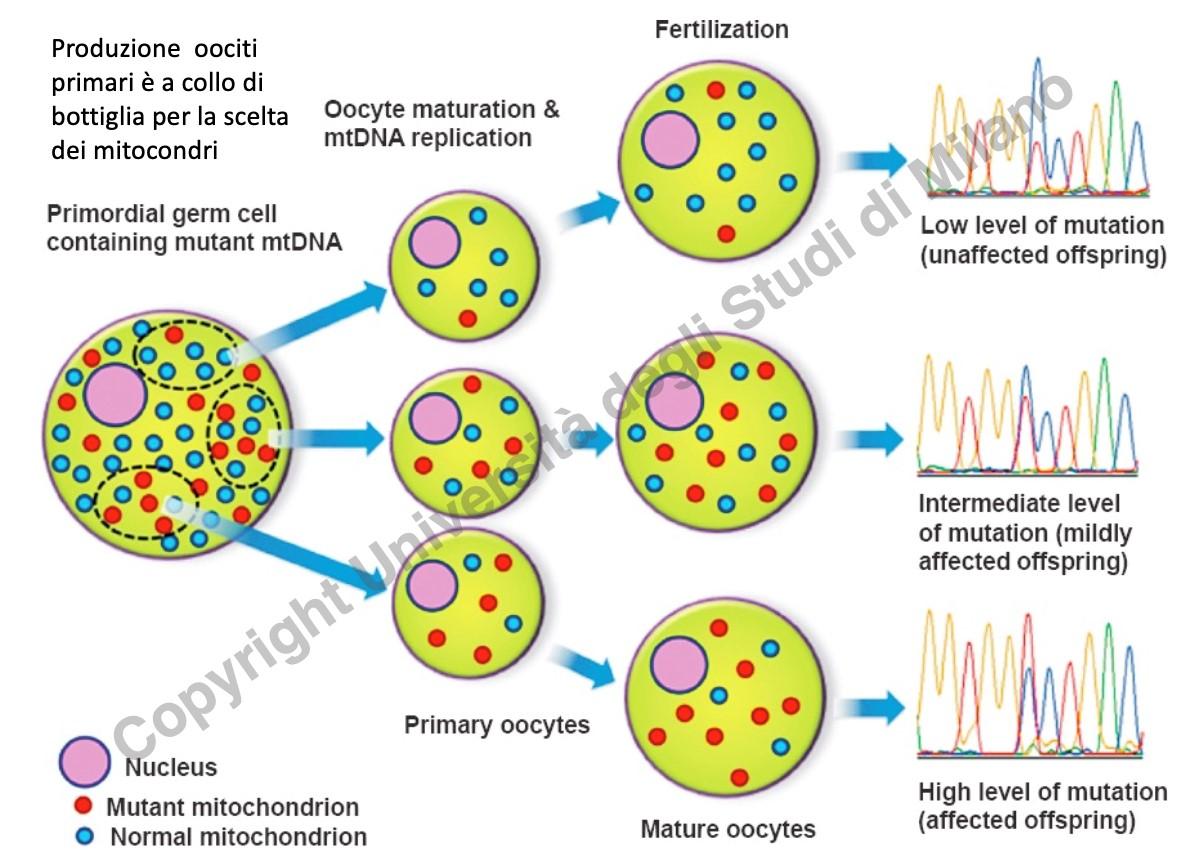
Questa fluttuazione fenotipica è regolata dal collo di bottiglia mitocondriale durante l’oogenesi:
- Nelle cellule germinali primordiali (oociti primordiali) si verifica una massiccia replicazione e amplificazione dei mitocondri.
- Nel passaggio a oocita primario avviene una contrazione numerica drastica dei mitocondri (collo di bottiglia): solo un piccolo campione casuale di mitocondri viene trasmesso alle cellule figlie.
- Durante la maturazione meiotica finale, questo campione ristretto viene nuovamente replicato. La proporzione finale di mtDNA mutato nell’oocita maturo fecondato dipenderà interamente dalla composizione casuale del campione iniziale passato attraverso il “collo di bottiglia”, determinando penetranza incompleta ed espressività variabile nella prole.
Quadro Clinico
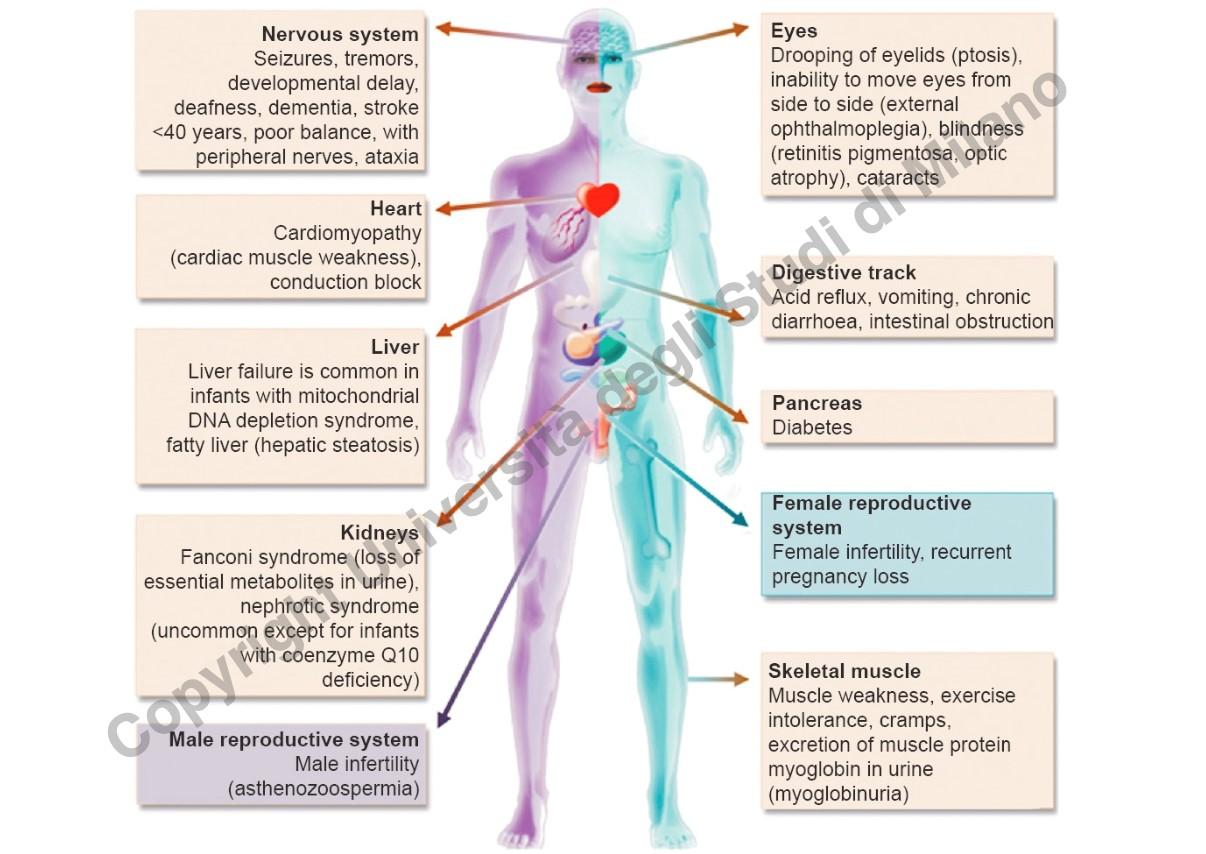
Le patologie del mtDNA colpiscono prevalentemente gli organi e i tessuti a elevato fabbisogno energetico (tessuti post-mitotici aerobici):
- Sistema Nervoso Centrale: Crisi epilettiche, tremori, ritardo dello sviluppo psicomotorio, sordità neurosensoriale, demenza, episodi simil-ictus (stroke-like) prima dei 40 anni, atassia.
- Apparato Oculare: Ptosi palpebrale (indicata talvolta come artrosi palpebrale), oftalmoplegia esterna progressiva (difficoltà di movimento oculare), cecità, cataratte.
- Apparato Cardiovascolare: Cardiomiopatia ipertrofica o dilatativa.
- Tessuto Muscolare Scheletrico: Debolezza muscolare (miopatia), crampi, mioglobinuria (danno cellulare con rilascio di mioglobina nelle urine).
- Organi Endocrini e Parenchimatosi: Compromissione di fegato e pancreas (diabete mitocondriale).
Età di Insorgenza
Possono manifestarsi a qualunque età, ma mostrano un picco di incidenza durante la pubertà (6-20 anni), fase caratterizzata da un marcato incremento del metabolismo basale e delle richieste energetiche dell’organismo.
Approccio Diagnostico
- Analisi dell’Albero Genealogico: Identificazione del pattern di trasmissione matrilineare o di casi sporadici a esordio clinico correlabile.
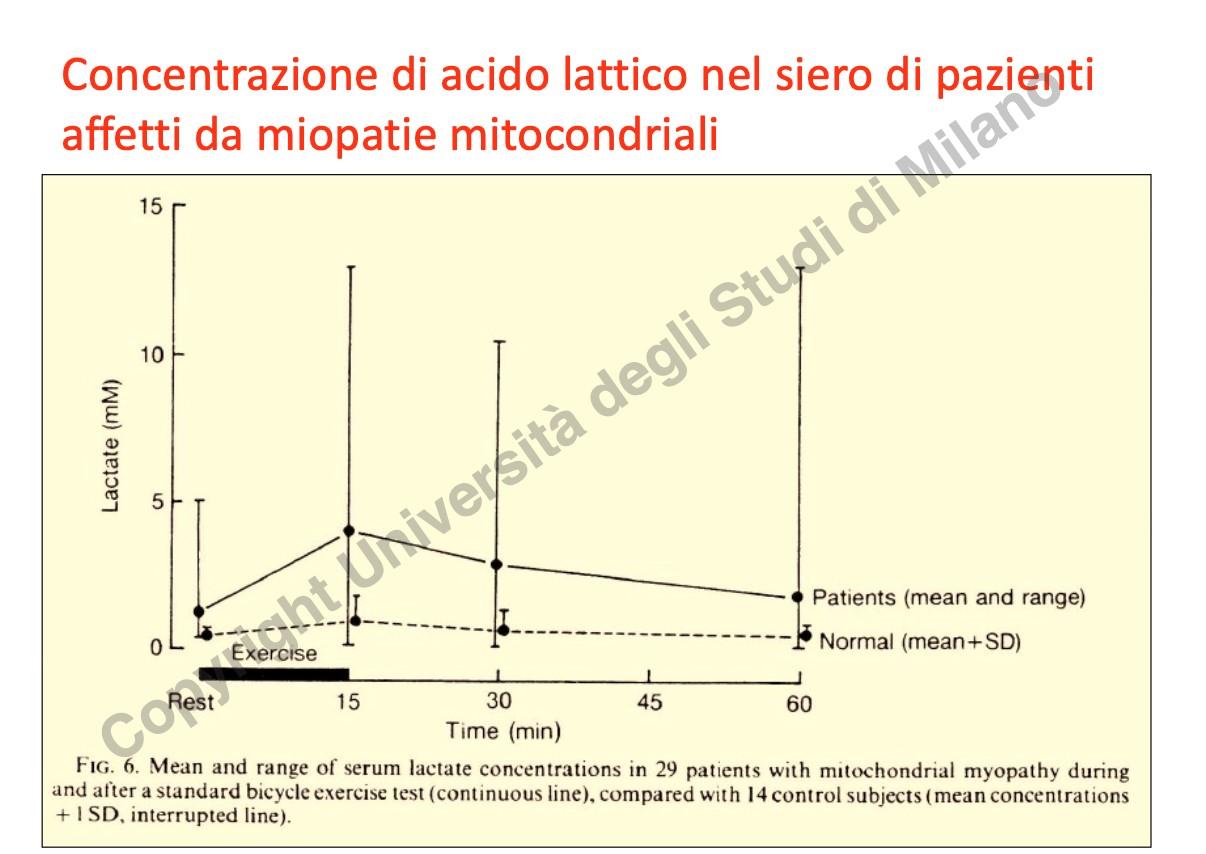
- Dosaggio dell’Acido Lattico: L’accumulo di lattato nel sangue e nel liquor (specialmente dopo sforzo fisico) indica uno shift verso la glicolisi anaerobia dovuto a deficit della catena respiratoria.
- Biopsia Muscolare: Evidenzia alterazioni istologiche caratteristiche (es. fibre rosse sfilacciate, ragged red fibers) e deficit biochimici dei complessi respiratori.
- Analisi Molecolare del mtDNA: Sequenziamento del mtDNA per identificare mutazioni puntiformi o riarrangiamenti.

Classificazione Clinico-Molecolare
Malattie di Classe I
Causate da alterazioni intrinseche del genoma mitocondriale (mtDNA):
- Mutazioni Puntiformi: Colpiscono i geni codificanti o i geni per tRNA/rRNA. Le mutazioni nei geni dei tRNA (es. nella MELAS o MERRF) sono particolarmente deleterie poiché bloccano globalmente la traduzione di tutte le 13 proteine codificate dal mitocondrio.
- Delezioni: Perdite di segmenti di mtDNA (da 1.3 a 8 kb). Sono prevalentemente eventi sporadici insorti nello zigote (non ereditati); determinano fenotipi gravi e spesso sterilità.
Malattie di Classe II
Causate da mutazioni del DNA nucleare che compromettono la funzione mitocondriale:
- Sindromi della Fosforilazione Ossidativa: Mutazioni nei geni nucleari codificanti subunità strutturali dei complessi respiratori (I, II, III, IV, V).
- Difetti nei Geni di Manutenzione: Mutazioni in proteine deputate alla replicazione e stabilità del mtDNA (es. polimerasi gamma, determinando delezioni multiple secondarie del mtDNA).
- Patologie da Effetti Secondari: Alterazioni in geni nucleari non direttamente correlati alla catena respiratoria ma che causano un danno mitocondriale secondario.
🔗 Collegamenti
- Ereditarietà Legata ai Cromosomi Sessuali — 🔄 confronto
- Albero Genealogico — 🔬 reperto diagnostico
- Gametogenesi — 🔗 stesso meccanismo / stessa via
- Donazione Mitocondriale — 💊 trattamento