Albero Genealogico
L’albero genealogico (o pedigree) è lo schema grafico delle relazioni familiari e dello stato clinico dei membri di una famiglia su più generazioni. Rappresenta lo strumento d’elezione per studiare la trasmissione di un carattere mendeliano (un carattere genico semplice la cui presenza o assenza dipende dal genotipo di un singolo locus).
Il pedigree permette di dedurre i genotipi individuali a partire dai fenotipi (sano vs affetto).
Razionale dell’Analisi Genealogica
Nello studio della genetica umana, l’albero genealogico è indispensabile per diversi motivi:
- Dimensione Familiare Ridotta: Le famiglie umane contemporanee sono piccole (in media nucleari, composte da genitori e 1-2 figli), il che limita la significatività statistica diretta delle segregazioni alleliche.
- Incroci non Controllati: Avvengono spesso tra portatori sani (eterozigoti) asintomatici. Per patologie autosomiche recessive diffuse (es. Fibrosi Cistica, con frequenza di portatori pari a 1 su 25 nei caucasici; o Anemia Falciforme nelle popolazioni africane), l’unione tra eterozigoti comporta il 25% di probabilità di generare prole affetta.
- Manifestazione dei Caratteri Dominanti: Le mutazioni patologiche dominanti si manifestano quasi esclusivamente in eterozigosi (
Aa). L’omozigosi dominante (AA) è estremamente rara e spesso letale in fase embrionale; pertanto, i soggetti affetti in pedigree dominanti sono da considerarsi eterozigoti.
Struttura e Convenzioni di Rappresentazione
L’albero genealogico è organizzato secondo una griglia bidimensionale standardizzata dal Pedigree Standardization Work Group (PSWG):
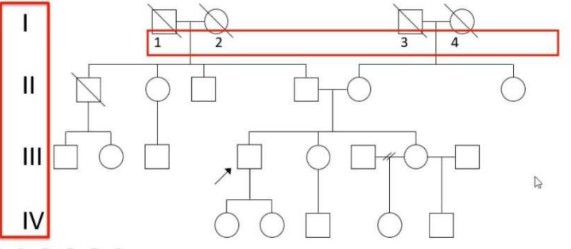
- Generazioni (Asse Verticale): Indicate con numeri romani (I, II, III, IV,…) in ordine decrescente (la generazione I rappresenta la più antica).
- Individui (Asse Orizzontale): Indicati all’interno di ciascuna generazione con numeri arabi (1, 2, 3, 4,…) da sinistra verso destra.
- Esempio: L’identificativo II-6 si riferisce al sesto individuo (da sinistra) della seconda generazione.
- Ordine di Nascita: I figli di una coppia vengono disposti da sinistra a destra in ordine cronologico di nascita, dal primogenito al più giovane.
- Disposizione dei Partner: Per convenzione, il partner maschile è posizionato a sinistra e quello femminile a destra.
I dettagli grafici sui simboli individuali, sulle linee parentali e sulla riproduzione assistita sono descritti nella Simbologia del Pedigree.
Protocollo di Raccolta Dati
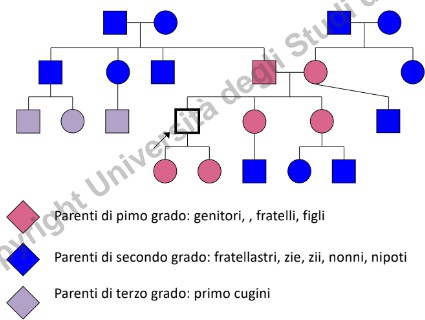
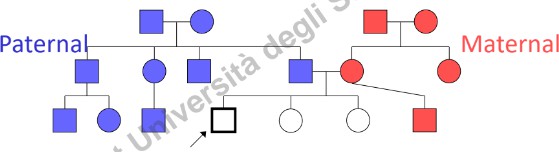
Per essere clinicamente utile, l’albero genealogico deve raccogliere dati dettagliati relativi ad almeno 3 generazioni (parenti di primo, secondo e terzo grado, sia del ramo materno sia di quello paterno).
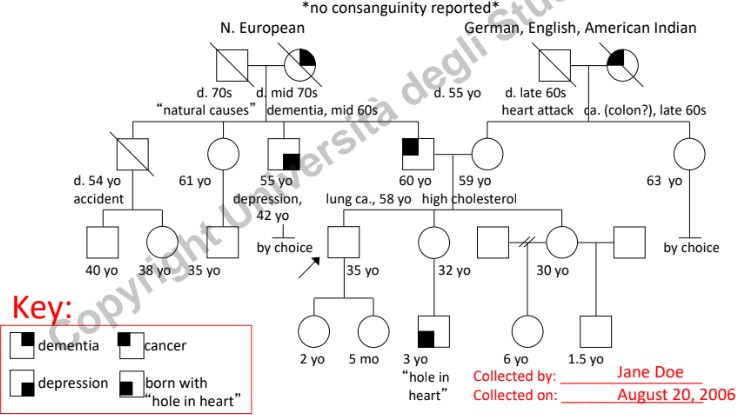
Soggetti Chiave da Identificare
- Consultando (o Probanda/o): L’individuo che richiede la consulenza genetica (indicato con una freccia).
- Probando: Il caso indice, ossia il primo individuo affetto che porta la famiglia all’attenzione del medico (indicato con una freccia che parte dalla lettera P).
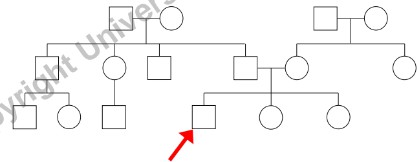
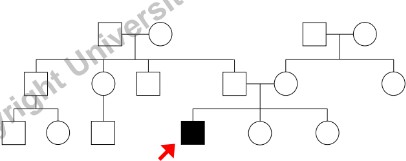
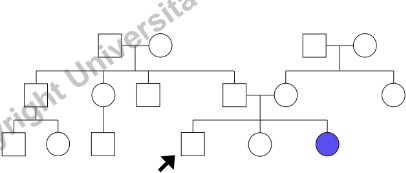
Informazioni da Raccogliere per Ogni Individuo
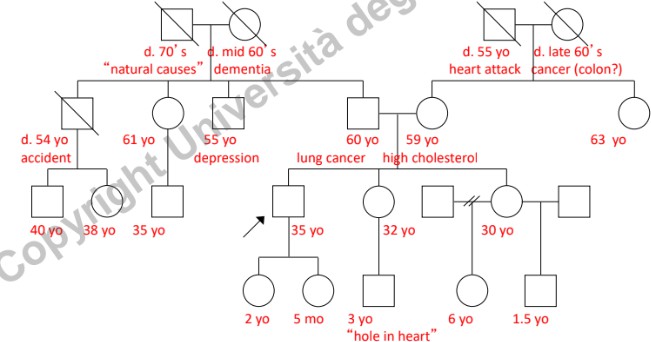
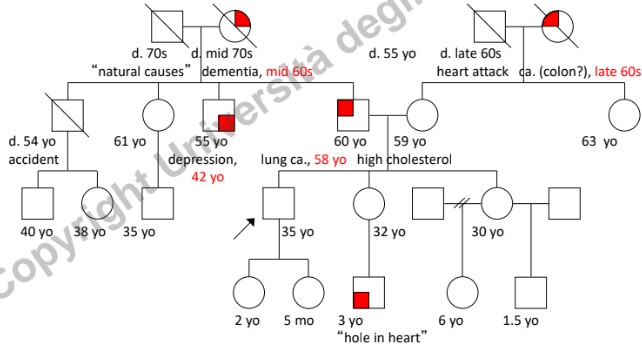
- Dati Anagrafici: Età attuale, data o anno di nascita. In caso di decesso, annotare età, anno e causa di morte.
- Dati Clinici: Diagnosi specifiche ed età all’esordio dei sintomi.
- Nota oncologica: Nei tumori a base genetica ereditari, l’esordio è tipicamente giovanile (sotto i 50-55 anni). L’insorgenza di neoplasie in età senile (>50 anni) è più frequentemente di natura sporadica (non ereditaria), legata all’accumulo di mutazioni somatiche nel tempo.
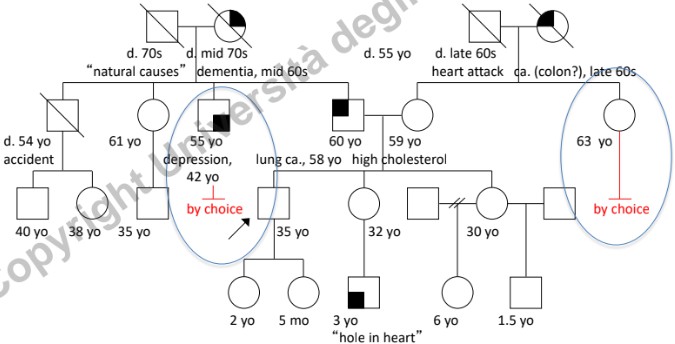
- Infertilità: Distinguere la scelta personale/cause non patologiche (es. vasectomia) dall’infertilità biologica (es. azoospermia, endometriosi severa). È fondamentale discriminare l’infertilità dall’abortività precoce dovuta a mutazioni letali che provocano il riassorbimento dell’embrione (spesso percepito dalla donna solo come un ritardo mestruale).
- Gravidanze e Complicanze: Registrare aborti spontanei (SAB), interruzioni volontarie di gravidanza (TOP), morti alla nascita (SB), gravidanze ectopiche (ECT), complicazioni emorragiche e preeclampsia (sofferenza fetale da ipertensione materna, generalmente non genetica).
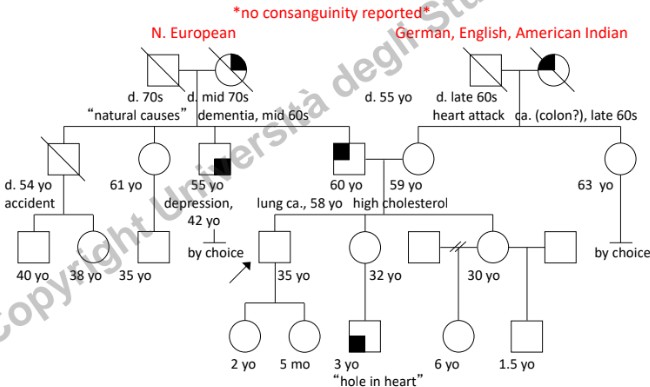
- Background Etnico e Provenienza: Registrare l’etnia dei quattro nonni biologici. Identificare l’appartenenza a popolazioni geograficamente o culturalmente “chiuse” (assenza di flussi migratori), poiché aumentano la frequenza di determinati alleli mutati (es. talassemia in alcune aree come il Lodigiano).
- Consanguineità: Segnalare unioni tra consanguinei (indicate con doppia linea), in quanto aumentano significativamente il rischio di omozigosi per alleli recessivi deleteri.
Anamnesi Specifica sul Probando
- Decorso Perinatale: Andamento della gravidanza, ecografie morfologiche (es. quarto mese), modalità del parto, presenza di ipossia neonatale e punteggio Apgar.
- Pietre Miliari dello Sviluppo: Ritardi nello sviluppo motorio, cognitivo o sensoriale (es. età di inizio della deambulazione, del linguaggio, della capacità visiva/uditiva e di relazione). I bambini con distrofia muscolare di Duchenne presentano anomalie precoci nel gattonamento e nel cammino.
- Dismorfismi e Anomalie Congenite: Presenza di tratti somatici atipici (es. impianto auricolare basso, rima palpebrale invertita). È necessario confrontare il fenotipo con quello dei genitori per distinguere variazioni familiari fisiologiche (“tratti ereditari benigni”) da veri dismorfismi sindromici.
- Fattori Ambientali: Esposizione della madre o del probando a sostanze chimiche, tossici lavorativi o farmaci teratogeni durante la gestazione.
- Storia Medica: Pregressi ricoveri, interventi chirurgici e terapie farmacologiche croniche.
Obiettivi di Valutazione della Trasmissione
L’albero genealogico mira a determinare a quale delle seguenti classi appartenga la trasmissione del carattere studiato:
1. Ereditarietà Monogenica (Mendeliana)
- Ereditarietà Autosomica Dominante (AD): Si manifesta in ogni generazione; colpisce maschi e femmine in egual misura.
- Ereditarietà Autosomica Recessiva (AR): Tende a saltare generazioni; frequente in caso di consanguineità.
- Ereditarietà X-Linked Dominante (XD): Le femmine sono colpite più frequentemente dei maschi; un padre affetto trasmette il carattere a tutte le figlie ma a nessun figlio maschio.
- Ereditarietà X-Linked Recessiva (XR): Colpisce prevalentemente i maschi; trasmessa da madri portatrici sane.
- Ereditarietà Y-Linked (Inesistenza di Malattie Y-Linked): Non si riscontrano pedigree ereditari di malattie genetiche a trasmissione Y-linked, in quanto le mutazioni sui pochi geni del cromosoma Y (es. spermatogenesi o differenziamento) provocano tipicamente azoospermia e sterilità primaria. Esiste tuttavia la trasmissione Y-linked di caratteri fenotipici non patologici (es. ipertricosi auricolare).
2. Ereditarietà Multifattoriale (Complessa)
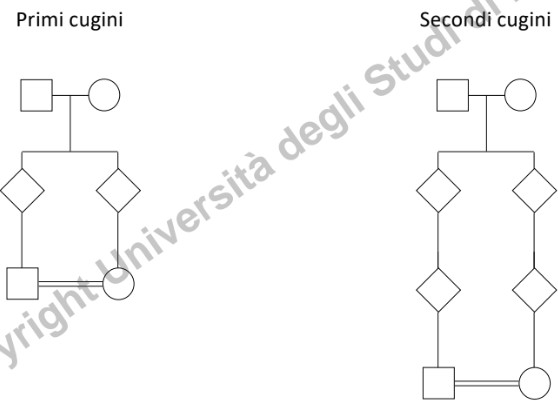
La segregazione non segue modelli mendeliani semplici. La ricorrenza familiare dipende dal grado di parentela: parenti stretti (primo grado) condividono una frazione maggiore di geni e quindi mostrano una suscettibilità più alta, che cala progressivamente nei parenti più distanti (es. cugini di primo grado).
3. Ereditarietà Mitocondriale (Citoplasmatica)
Trasmissione esclusivamente materna. Le madri affette trasmettono il carattere a tutta la progenie (sia maschi sia femmine); i maschi affetti manifestano la condizione ma non la trasmettono a nessun figlio, poiché i mitocondri dello zigote derivano unicamente dal citoplasma dell’ovocita.
4. Ereditarietà Cromosomica
Legata ad alterazioni nel numero di cromosomi (es. trisomia 21 nella Sindrome di Down), spesso dovute a errori di non-disgiunzione meiotica e caratterizzate da un rischio di ricorrenza basso ma correlato all’età materna.
(Integrazione da: sbobina 15)
Regole di Calcolo del Rischio nei Pedigree
L’analisi probabilistica degli alberi genealogici richiede l’applicazione di principi mendeliani integrati da regole matematiche per stimare il rischio di trasmissione di caratteri patogeni.
1. Calcolo del Portatore Sano per Malatte Autosomiche Recessive (Regola del 2/3)
Nelle patologie autosomiche recessive, la probabilità che un individuo sano sia portatore eterozigote (Aa), avendo un fratello o sorella affetto (aa), è pari a 2/3 (e non a 2/4 o 1/2).
- Razionale: I genitori di un individuo affetto (
aa) devono essere necessariamente portatori sani (AaxAa). La segregazione mendeliana prevede:AA,Aa,aa. Sapendo a priori che l’individuo in esame è fenotipicamente sano, si esclude la classe omozigote recessiva (aa) dal denominatore. Lo spazio campionario si riduce a 3 combinazioni possibili, di cui 2 sono portatori (Aa), determinando una frazione di . - Esempio: Se due fidanzati sani hanno ciascuno una sorella affetta, la probabilità che il loro primo figlio sia affetto è:
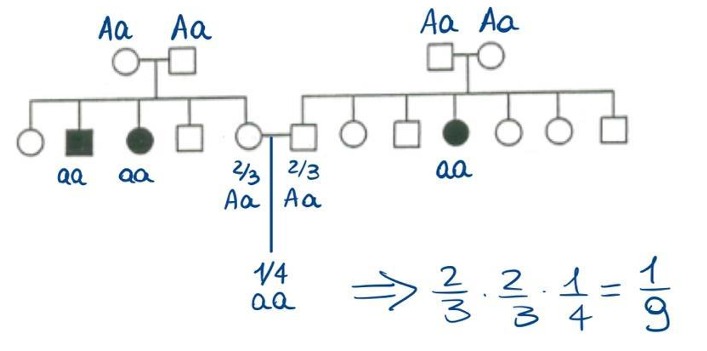
2. Consanguineità e Trasmissione Transgenerazionale
Nelle unioni tra consanguinei, la probabilità che un allele mutato raro presente in un antenato comune venga ereditato in omozigosi (causando la patologia recessiva nella prole) si calcola tracciando le generazioni:
- Regola del dimezzamento: La probabilità che un genitore eterozigote trasmetta l’allele mutato a un figlio è pari a . Ad ogni passaggio generazionale (salita verso l’antenato comune e discesa verso il partner consanguineo), la probabilità si dimezza di un fattore .
- Esempio (unione tra cugini con antenato comune portatore): Se una trisavola comune (I-2) è portatrice eterozigote (
Aa), la probabilità che il partner V-1 sia portatore è (4 passaggi generazionali). Analogamente, per il partner V-2 la probabilità è . La probabilità che il loro primo figlio sia affetto (aa) è:Transclude of 15---28-10-24---genetica-mz-img20.emf
3. Calcolo Combinato di Loci Indipendenti (Regola del Prodotto)
Per calcolare il rischio contemporaneo relativo a caratteri o patologie localizzati su geni indipendenti (autosomi diversi o cromosomi sessuali), si applica la regola del prodotto (moltiplicazione delle probabilità dei singoli eventi):
- Esempio (Fibrosi Cistica e PKU): Se due genitori sono portatori sani per entrambe le malattie (
Pp FfxPp Ff), la probabilità che il figlio manifesti la fibrosi cistica (ff, probabilità ) ma NON la fenilchetonuria (P-, ovvero sano o portatore di PKU, probabilità ) è:
4. Condizionamento da Informazione Nota
Nel calcolo delle probabilità, se un’informazione è fornita come certezza clinica (es. “qual è la probabilità che il figlio maschio sia daltonico” o “sapendo che la figlia è sana”), tale probabilità condizionante non deve essere moltiplicata nel calcolo finale.
- Esempio: Se si chiede la probabilità che “un figlio maschio” sia affetto da emofilia da madre portatrice, la probabilità è (la probabilità di ereditare la X mutata), senza moltiplicare per (probabilità che sia maschio), in quanto il sesso del nascituro è già stabilito dalla domanda.
5. Penetranza Incompleta e Letalità Omozigote
- Penetranza incompleta: Se una patologia dominante ha una penetranza (es. ), la probabilità di avere un figlio affetto è data dal prodotto tra la probabilità genotipica (es. se un genitore è
Aa) e la penetranza (es. ): - Letalità omozigote precoce: Nelle patologie dominanti letali in utero (es. acondroplasia o sindrome di Marfan), l’omozigote dominante
AAnon sopravvive. Di conseguenza, l’incrocio tra due eterozigotiAaxAapresenta uno spazio campionario dei nati vivi ridotto a 3 combinazioni (AAesclusa):- Probabilità di affetto (nato vivo,
Aa) = - Probabilità di sano (nato vivo,
aa) =
- Probabilità di affetto (nato vivo,
🔗 Collegamenti
- Anamnesi Familiare — ⬆️ causa
- Malattie Genetiche — ⬆️ causa
- Simbologia del Pedigree — 🔗 stesso meccanismo / stessa via
- Sindrome di Down — 🔬 reperto diagnostico
- Fibrosi Cistica — 🔬 reperto diagnostico
- Anemia Falciforme — 🔬 reperto diagnostico
- Gemelli — 📋 fa parte di
- Ereditarietà Autosomica Dominante — 📋 fa parte di
- Ereditarietà Autosomica Recessiva — 📋 fa parte di
- Ereditarietà Legata ai Cromosomi Sessuali — 📋 fa parte di
- Ereditarietà X-Linked Recessiva — 📋 fa parte di
- Ereditarietà X-Linked Dominante — 📋 fa parte di
- Ereditarietà Y-Linked — 📋 fa parte di