Sindrome di Down
La sindrome di Down è un’alterazione cromosomica causata dalla presenza di una tripla copia del materiale genetico del cromosoma 21 (trisomia 21) nel cariotipo. La sua eziologia cromosomica fu scoperta nel 1959 da Lejeune J..
Eziologia e Meccanismi Genetici
La trisomia 21 può originare da tre differenti genotipi:
1. Non-Disgiunzione Meiotica / Trisomia Libera (95% dei casi)
Il cariotipo presenta tre cromosomi 21 liberi in sovrannumero (), causati da un errore di disgiunzione (mancata separazione dei cromosomi omologhi o dei cromatidi fratelli) durante la divisione meiotica, principalmente di origine materna.
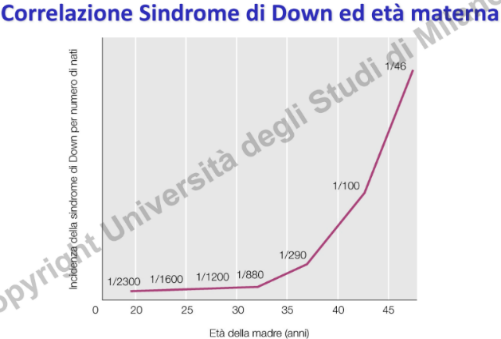
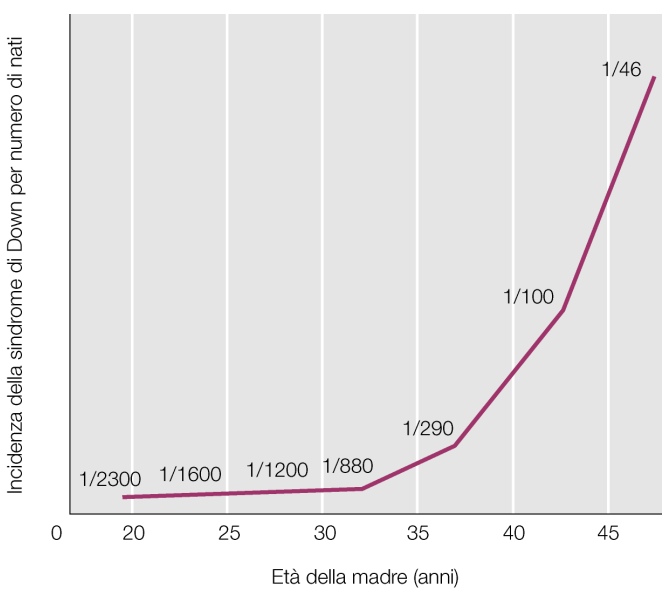
- Correlazione con l’Età Materna: Esiste una stretta correlazione statistica tra l’aumento dell’età della madre e l’insorgenza della trisomia 21. Questo fenomeno è legato al blocco prolungato degli oociti in Profase I. La gametogenesi femminile inizia durante la vita fetale (II trimestre di gestazione), per poi bloccarsi alla nascita in profase I. Durante questo lungo periodo di standby, che si protrae fino alla pubertà, il numero di gameti femminili diminuisce significativamente. A partire dalla pubertà, una o poche cellule al mese riprendono la meiosi, completando la meiosi II esclusivamente in caso di fecondazione. Con l’aumentare degli anni in questo stato di arresto, l’apparato di disgiunzione cromosomica accumula difetti, aumentando il rischio di errori meiotici (solitamente in meiosi I).
- Screening Prenatale: Viene proposto dal sistema sanitario di routine alle donne oltre i 35 anni. Può essere indirizzato anche dal riscontro ecografico di anomalie come la translucenza nucale (uno spessore maggiore della nuca fetale riscontrabile nei pazienti Down).
- Tracciamento dell’Origine: È possibile determinare quale genitore ha trasmesso il cromosoma extra e in quale fase meiotica sia avvenuto l’errore tramite l’analisi di marcatori polimorfici come i Microsatelliti.
2. Traslocazione Robertsoniana Sbilanciata (3% dei casi)
L’eccesso di materiale genetico del cromosoma 21 è dovuto alla fusione del braccio lungo del cromosoma 21 con quello di un altro cromosoma acrocentrico (frequentemente il cromosoma 14). Il cariotipo è 46, XX/XY, rob(14;21)(q10;q10), +21.
- Origine: Questa anomalia strutturale origina spesso da un genitore portatore sano di una Traslocazione Robertsoniana bilanciata (avente 45 cromosomi, ma fenotipo normale).
- Rischio di Ricorrenza: A differenza della non-disgiunzione (evento sporadico legato all’età), il portatore di traslocazione bilanciata presenta un rischio elevato e ricorrente di trasmettere un gamete sbilanciato, determinando la nascita di figli affetti da sindrome di Down indipendentemente dall’età materna.
(Integrazione da: sbobina 23)
- Rischio in base al sesso del genitore portatore di rob(14;21):
- Madre portatrice: rischio del 10-15% di prole con sindrome di Down.
- Padre portatore: rischio dell’1-2%.
- Traslocazione rob(21;21): Se la traslocazione robertsoniana coinvolge entrambi i cromosomi 21 dello stesso genitore, il rischio di prole affetta è del 100%: alla meiosi si formano esclusivamente gameti disomici (con il cromosoma derivativo contenente entrambi i bracci q del 21) o nullisomici (senza cromosoma 21). I gameti disomici, una volta fecondati, producono zigoti trisomici per il 21; i nullisomici producono zigoti monosomici per il 21, condizione letale.
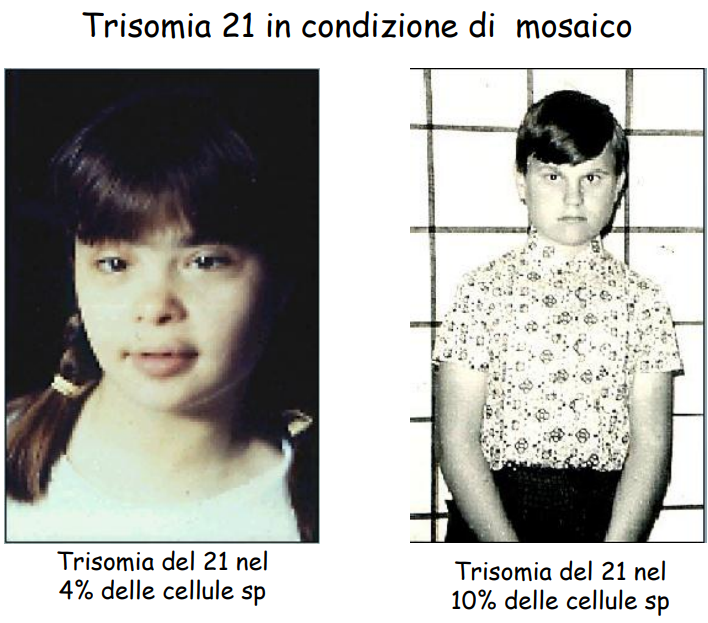
3. Mosaicismo Mitotico Post-zigotico (1.2% dei casi)
Il paziente presenta la coesistenza di linee cellulari diploidi normali () e di linee cellulari trisomiche (), causate da una non-disgiunzione mitotica durante le prime divisioni dello zigote. Il fenotipo clinico è estremamente variabile in base a quanto precocemente sia avvenuto l’errore e a quali tessuti/foglietti embrionali siano coinvolti.
Basi Biologiche della Vitalità
La sindrome di Down rappresenta la trisomia autosomica più frequente e vitale nella popolazione umana.
- Dimensioni del Cromosoma: Il cromosoma 21 è il più piccolo del cariotipo umano (circa 46,7 Mb) e contiene in percentuale un numero di geni notevolmente inferiore rispetto ad altri cromosomi.
- Dosaggio Genico: Poiché il numero assoluto di geni coinvolti nella tripla copia è ridotto, lo sbilanciamento di dosaggio genico che ne deriva è compatibile con la vita dell’individuo, pur determinando complicanze cliniche, fisiche e cognitive di grado variabile. Al contrario, trisomie che coinvolgono cromosomi più grandi e densamente popolati di geni risultano letali nelle prime fasi dello sviluppo embrionale.
(Sezione espansa con: sbobina 19)
(Integrazione da: sbobina 22)
Rilevanza Epidemiologica
L’incidenza complessiva alla nascita è di circa 1 su 850 nati vivi (è la trisomia autosomica più comune). Tuttavia, l’aborto spontaneo esercita una fortissima selezione naturale: solo il 20% dei concepimenti con trisomia 21 sopravvive fino al termine della gravidanza. Questa frequenza naturale alla nascita è uniforme in tutte le popolazioni, differendo solo in base alla disponibilità ed al ricorso a programmi di diagnosi prenatale.
Quadro Clinico e Fenotipo
Sebbene la sindrome sia compatibile con la sopravvivenza post-natale, presenta caratteristiche cliniche peculiari e complicanze organiche sistemiche:
- Sviluppo Neurologico: Alla nascita si rilevano ipotonia (ridotto tono muscolare) e ritardo psico-motorio e cognitivo di grado variabile.
- Segni Dismorfici: Bassa statura (corporatura bassa e tozza), piega palmare singola (linea simiana), nuca piatta, microcefalia. Viso rotondo e piatto, macroglossia (lingua sporgente), taglio palpebrale obliquo (fessure palpebrali orientate verso l’alto), orecchie piccole.
- Malformazioni Organiche: Nel 50% dei casi sono presenti malformazioni cardiache congenite. Possono presentarsi anche atresia esofagea o stenosi duodenale.
- Patologie Secondarie: Deficit del sistema immunitario con aumentata suscettibilità alle infezioni batteriche; tendenza a sviluppare leucemia acuta (circa 1% dei casi), ipotiroidismo e diabete mellito.
Consulenza Genetica
Nel caso della presenza di un figlio affetto da trisomia 21 libera (non da traslocazione):
- L’evento di non-disgiunzione è considerato sporadico e non ereditario.
- Il rischio di ricorrenza per gravidanze future della stessa coppia è molto basso, stimato in un incremento di appena l’1% rispetto al rischio generico per l’età materna (aumento dovuto alla remota possibilità di mosaicismo germinale gonadico in uno dei genitori).
- Il rischio per la progenie di altri parenti (inclusi i fratelli sani del soggetto affetto) non è aumentato ed è pari a quello della popolazione generale.
🔗 Collegamenti
- Cariotipo — 🔬 reperto diagnostico
- Gametogenesi — ⬆️ causa
- Cromosomi — 🔗 stesso meccanismo / stessa via
- Anomalie Cromosomiche — ⬆️ causa
- Traslocazione Robertsoniana — ⬆️ causa