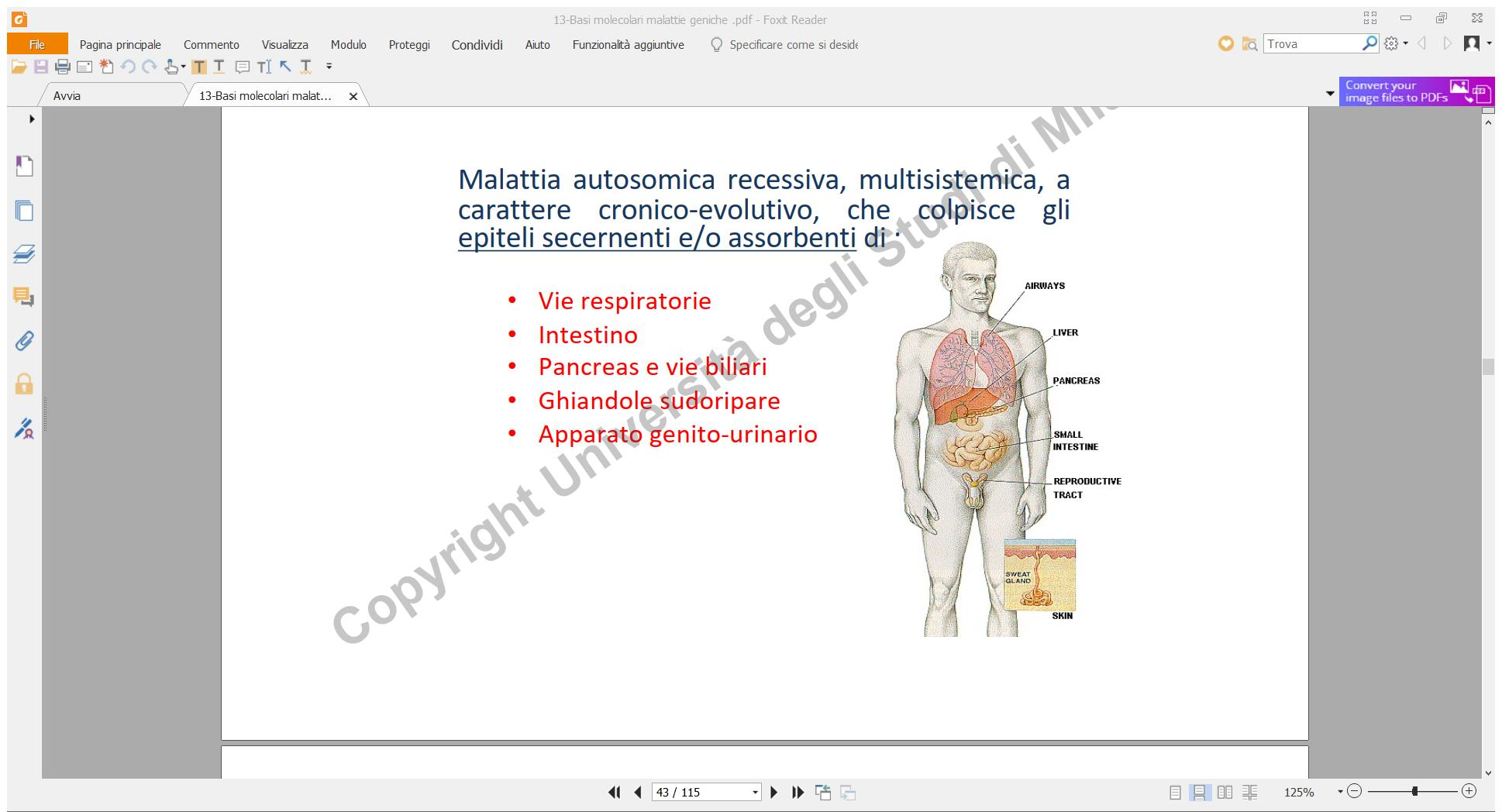
Fibrosi Cistica
La fibrosi cistica è una grave patologia genetica multisistemica a trasmissione autosomica recessiva, caratterizzata da un’elevata frequenza nella popolazione caucasica. Colpisce principalmente gli epiteli secernenti e ciliati dell’organismo.
Eziopatogenesi, Trasmissione ed Epidemiologia
La patologia è causata da mutazioni a carico del gene CFTR (situato sul braccio lungo del cromosoma 7), che codifica per un canale ionico transmembrana regolatore del flusso del cloro. La disfunzione del canale impedisce il corretto trasporto idrosalino, determinando la secrezione di muco patologicamente denso e disidratato.
- Epidemiologia: Colpisce prevalentemente la popolazione caucasica (Europa, Stati Uniti e Australia). In Italia, l’incidenza è di circa 1 su 2438 nati vivi.
- Frequenza dei Portatori: I soggetti eterozigoti sono portatori sani asintomatici. La frequenza dei portatori sani nella popolazione caucasica è stimata essere di circa 1 su 20 o 1 su 25.
- Vantaggio dell’Eterozigote: L’elevata prevalenza della patogenesi nella popolazione caucasica è legata a una selezione positiva degli eterozigoti (carrier). La parziale riduzione della conduttanza al cloro proteggeva i portatori sani dalla disidratazione e dalla perdita letale di elettroliti durante le epidemie storiche di colera e febbre tifoide (causata da Salmonella typhi).
- Consanguineità: Eleva notevolmente il rischio di accoppiamento tra due portatori sani, incrementando la probabilità di trasmettere la malattia alla progenie.
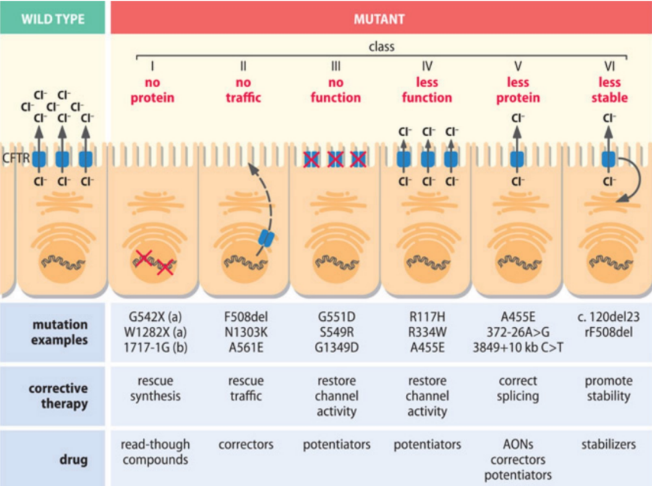
- Eterogeneità Mutazionale: Il gene presenta oltre 2000 mutazioni differenti. La gravità del fenotipo dipende dalla specifica combinazione degli alleli ereditati (eterogeneità allelica/genotipica).
Fisiopatologia e Presentazione Diagnostica (Test del Sudore)
La sintomatologia tipica del bambino affetto comprende:
- Deficit Staturo-Ponderale: Scarso accrescimento corporeo (percentile di crescita molto basso, es. 3° percentile) dovuto a malnutrizione per malassorbimento lipidico e vitaminico (mancata assimilazione delle vitamine liposolubili).
- Manifestazioni Intestinali: Diarrea frequente, coliche addominali e steatorrea (feci maleodoranti, lucide e ricche di grassi) causate dall’insufficienza del pancreas esocrino.
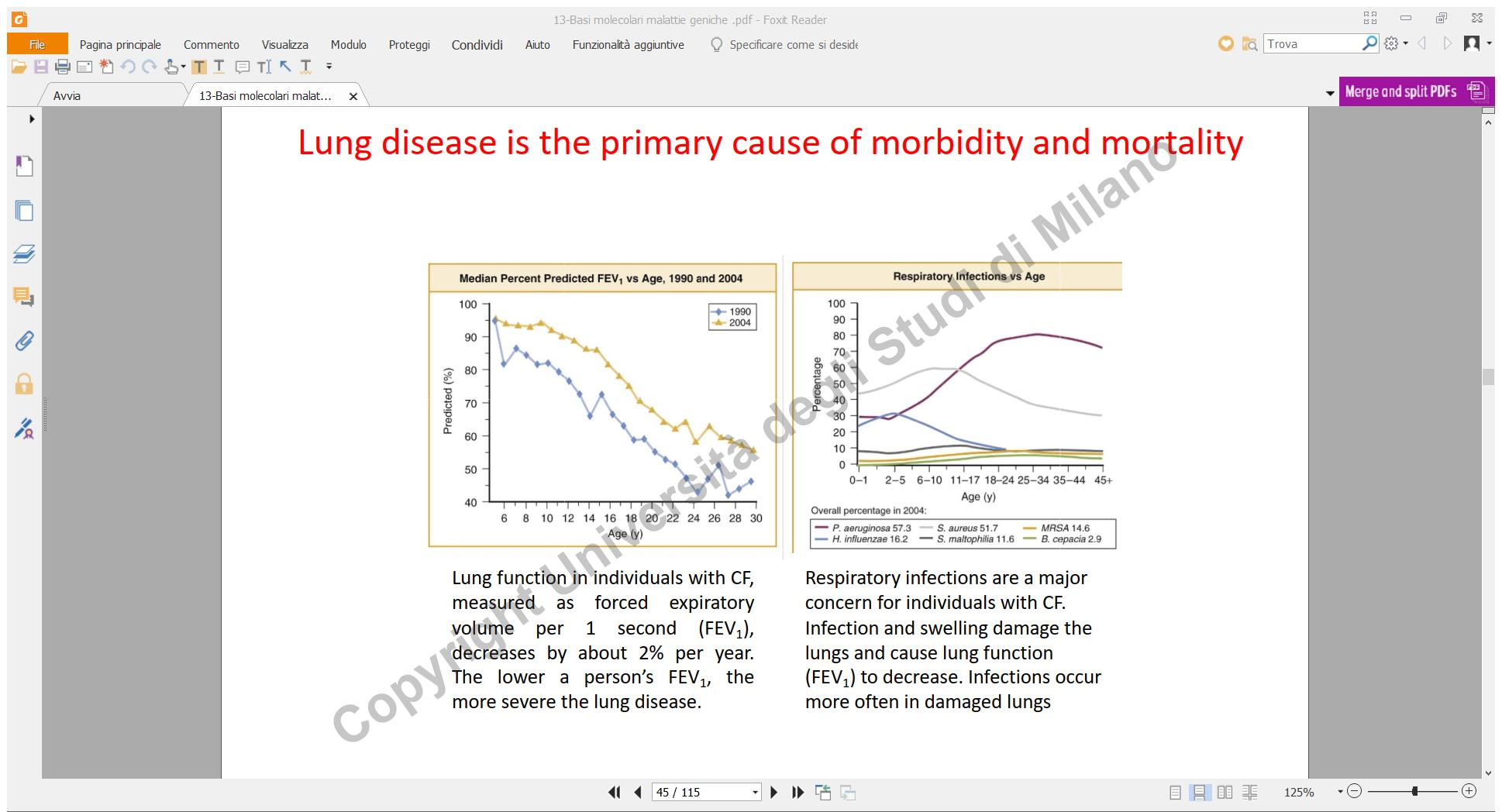
- Manifestazioni Respiratorie: Bronchiti e polmoniti recidivanti a causa del muco denso che ostruisce le vie aeree, bloccando il movimento delle ciglia epiteliali e favorendo infezioni batteriche croniche.
- Test del Sudore: La cute del paziente si presenta marcatamente salata. La diagnosi clinica standard si effettua a partire dal primo mese di vita tramite stimolazione della sudorazione con pilocarpina (mediante ionoforesi) e successiva misurazione della concentrazione di cloro e sodio nel sudore.
- Fisiologia del test: Nei soggetti sani, la ghiandola sudoripara filtra il plasma a livello della porzione convoluta. Per prevenire squilibri elettrolitici sistemici, il dotto sudoriparo riassorbe attivamente il sale: a questo livello, il canale CFTR lavora in senso inverso rispetto al polmone, riassorbendo il cloro all’interno della cellula (e trainando passivamente il sodio).
- Valori diagnostici: Nei soggetti normali, la concentrazione di sodio e cloro nel sudore è di circa 10-15 mEq/L. In caso di fibrosi cistica, l’assenza di CFTR funzionante impedisce il riassorbimento del cloro dal dotto; il cloro e il sodio rimangono nel sudore, determinando concentrazioni diagnostiche elevate, pari a circa 90 mEq/L.
Classificazione Clinica e Geni Modificatori
La correlazione tra genotipo e fenotipo non è sempre diretta a causa dell’influenza di geni modificatori e di fattori ambientali (es. inquinamento atmosferico urbano vs aria salubre di montagna).
- Apparato Pancreatico: Presenta una forte correlazione genotipo-fenotipo. La presenza di due mutazioni severe (Classi I, II, III) correla costantemente con la perdita totale della funzione pancreatica esocrina (insufficienza pancreatica).
- Apparato Respiratorio: La gravità clinica e la velocità di declino polmonare sono scarsamente correlate al genotipo CFTR, poiché fortemente modulate da geni modificatori extralocali. Polimorfismi a livello del gene TGFb1 (Transforming Growth Factor beta 1), ad esempio, possono determinare aplotipi protettivi che preservano più a lungo la funzionalità respiratoria e riducono la suscettibilità alle infezioni.
- Apparato Intestinale: Polimorfismi specifici possono predisporre i neonati affetti a ileo da meconio o a manifestazioni gravi di megacolon tossico, dovuti all’ostruzione da parte di feci neonatali eccessivamente compatte.
1. Forma Classica
Forma severa e multiorgano caratterizzata da:
- Apparato Respiratorio: Ostruzione mucosa polmonare progressiva con sinusite e infezioni batteriche croniche. Il tessuto polmonare viene gradualmente sostituito da tessuto fibrotico (fibrosi polmonare), con insufficienza respiratoria che storicamente richiedeva l’ausilio del polmone d’acciaio.
- Apparato Digerente ed Epatobiliare: Occlusione dei dotti biliari e dei dotti pancreatici esocrini. L’autodigestione pancreatica distrugge gli acini esocrini e può estendersi al pancreas endocrino, causando diabete secondario.
- Apparato Riproduttivo Maschile: Ostruzione e atrofia dei dotti deferenti che causa Assenza Congenita dei Vasi Deferenti (CAVD/CBAVD) e azoospermia.
2. Forma Non Classica (Meno Grave)
Caratterizzata da sufficienza pancreatica (pancreas esocrino funzionante). La sintomatologia respiratoria è più lieve o a insorgenza tardiva, ma permane costante la CAVD con infertilità maschile.
Gestione Terapeutica
- Terapia di Supporto: Somministrazione quotidiana di enzimi pancreatici sostitutivi prima dei pasti, monitoraggio dello sviluppo ponderale, vitamine liposolubili e fisio-kinesiterapia respiratoria per facilitare l’espettorazione del muco.
- Infezioni da Pseudomonas aeruginosa: Colonizzazione cronica delle vie aeree da parte di ceppi batterici multiresistenti agli antibiotici. Si utilizzano terapie antibiotiche aggressive e si studia l’impiego sperimentale di fagi naturali (virus batteriofagi) in grado di infettare e lisare selettivamente lo Pseudomonas.
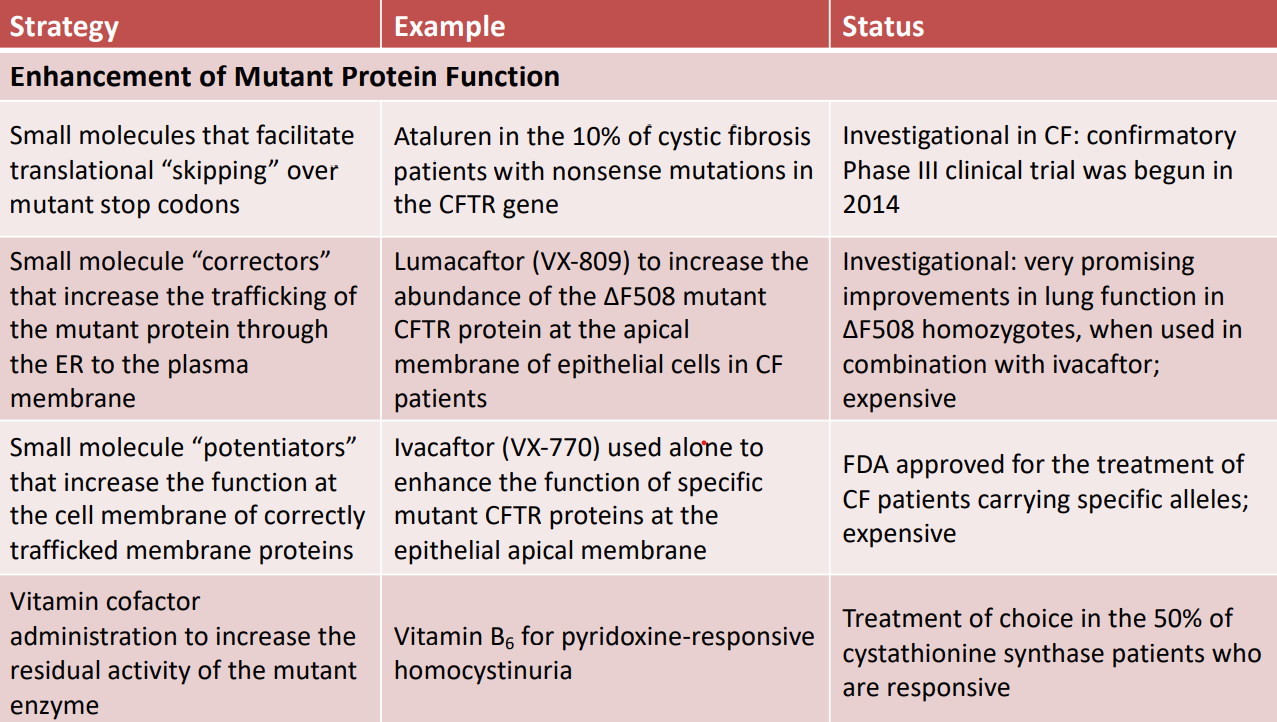
- Farmaci Modulatori (Terapie di Nuova Generazione): Molecole a basso peso molecolare (small molecules) destinate a correggere o potenziare la funzionalità del canale. Si suddividono in tre classi principali, spesso derivate dallo screening di librerie chimiche (che garantisce ridotti effetti collaterali e rapidità di identificazione):
- Agenti read-through: Permettono il superamento di codoni di stop prematuri (mutazioni nonsense). L’antibiotico amminoglicoside Ataluren (in trial clinico di fase III) si lega al ribosoma e inserisce un amminoacido non specifico al posto dello stop codon, permettendo il completamento della traduzione. Questa mutazione interessa solo il 10% dei pazienti con fibrosi cistica.
- Correttori: Intervengono sulle alterazioni del ripiegamento tridimensionale (folding). Nella mutazione (la più comune, presente nel 70% dei pazienti, appartenente alla Classe II), la proteina mal ripiegata non viene glicosilata e viene degradata prima di raggiungere la superficie cellulare. I correttori mirano a favorire il corretto ripiegamento e la glicosilazione, sebbene i trial clinici condotti con i soli correttori abbiano mostrato scarso successo.
- Potenziatori: Aumentano la funzione residua del canale (favorendo l’apertura del poro o aumentandone la conduttanza). Agiscono nelle mutazioni di Classe III e IV, a condizione che il canale sia già presente sulla membrana plasmatica.
- Terapia combinata: L’associazione clinica di correttori e potenziatori per i pazienti eterozigoti/omozigoti mostra risultati clinici nettamente superiori rispetto all’uso dei singoli farmaci modulatori.
- Terapia Genica (Adenovirus): Storicamente tentata veicolando il gene CFTR wild-type tramite vettori adenovirali in spray nasali. L’approccio è fallito poiché l’adenovirus stimolava una severa e massiva risposta infiammatoria polmonare nei pazienti, peggiorando il quadro clinico.
- Trapianto Polmonare: Rappresenta l’opzione terapeutica di ultima istanza per l’insufficienza respiratoria allo stadio terminale.
🔗 Collegamenti
- Seconda Legge di Mendel — ⬆️ causa
- Ereditarietà Autosomica Recessiva — 📋 fa parte di
- Eterogeneità Genetica — 📋 fa parte di
- Classificazione Funzionale delle Mutazioni — 🔗 stesso meccanismo / stessa via
- CFTR — ⬆️ causa (locus genico)
- Assenza Congenita dei Vasi Deferenti — ⬇️ conseguenza
- Ereditarietà Autosomica Dominante — 🔄 confronto (in relazione alle eccezioni omozigotiche o dominanti)
(Sezione espansa con: sbobina 13) (Sezione espansa con: sbobina 14) (Sezione espansa con: sbobina 18)