Approcci Terapeutici alle Malattie Genetiche
Gli approcci correttivi per il trattamento delle malattie genetiche (monogeniche) si applicano a differenti livelli biologici e clinici, muovendosi da una scala microscopica a una macroscopica. Le patologie complesse (poligeniche), a causa della loro elevata complessità e multifattorialità, non sono trattabili con queste metodologie e beneficiano esclusivamente di interventi preventivi.
Livelli di Intervento
Il trattamento può essere impostato intervenendo sui diversi passaggi del flusso dell’informazione genica e metabolica:
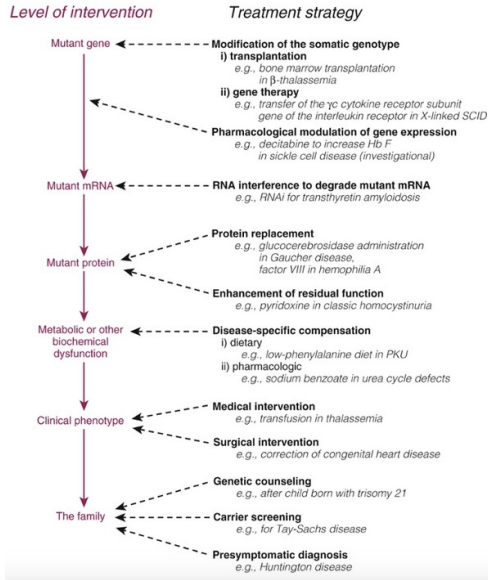
- Gene: Inserimento di una copia corretta del gene in presenza di mutazioni loss-of-function (es. terapia genica) o sostituzione di cellule staminali difettive (es. trapianto di midollo osseo nella Beta-Talassemia).
- RNA: Inibizione dell’espressione dell’allele mutato dominante tramite tecniche di RNA interference (RNAi) o modulazione dello splicing (es. oligonucleotidi antisenso o ASO).
- Proteina: Somministrazione della proteina wild-type prodotta in vitro (terapia enzimatica sostitutiva) o molecole volte a ripristinare la conformazione e la funzionalità della proteina endogena alterata.
- Metabolismo: Intervento sui pathway biochimici per evitare l’accumulo di metaboliti tossici o sopperire alla carenza di prodotti a valle del blocco enzimatico.
- Clinico: Gestione delle complicanze macroscopiche mediante chirurgia correttiva (es. labbro leporino) o terapie di supporto (es. emotrasfusioni).
- Familiare: Counseling genetico preventivo e identificazione precoce di soggetti portatori sani o presintomatici.
Interventi Preventivi
Rappresentano lo strumento principale per ridurre l’incidenza di gravi patologie e comprendono:
- Diagnosi prenatale: Indagini svolte in utero quali amniocentesi, villocentesi ed ecografia morfologica (eseguita nel secondo trimestre di gravidanza per valutare lo sviluppo degli organi).
- Fecondazione in vitro con diagnosi preimpianto: In coppie di portatori sani di malattie recessive (rischio di procreare figli affetti pari al 25%), permette di analizzare geneticamente gli embrioni e selezionare esclusivamente quelli non affetti o non portatori.
- Screening presintomatico: Identificazione precoce dei portatori di mutazioni per patologie ad esordio tardivo (es. Corea di Huntington) al fine di pianificare interventi terapeutici tempestivi o scelte riproduttive informate.
Strategie per Deficit Genetici (Recessive vs Dominanti)
L’approccio terapeutico differisce radicalmente in base al meccanismo patogenetico molecolare:
- Malattie recessive: Caratterizzate da perdita di funzione della proteina (loss-of-function). La terapia d’elezione è la augmentation therapy (terapia di aumento), che mira a ripristinare o supplementare la funzione biologica persa.
- Malattie dominanti (gain-of-function): Caratterizzate dalla presenza di alleli dominanti negativi o che conferiscono proprietà tossiche. Poiché l’editing genomico diretto sul DNA non è ancora utilizzabile in clinica per motivazioni etiche e trial non codificati, si interviene a livello dell’RNA tossico, con piccole molecole (small molecules) o con anticorpi monoclonali biologici per bloccare selettivamente la proteina mutata. Nelle anomalie congenite strutturali si ricorre alla chirurgia correttiva.
Approccio Metabolico
Consiste nella manipolazione biochimica del flusso metabolico alterato. Rappresenta il 28% di tutte le strategie terapeutiche applicate alle malattie genetiche.
1. Riduzione del Substrato (Substrate Reduction)
Consiste nella rimozione o forte riduzione dietetica del precursore a monte del blocco enzimatico per evitarne l’accumulo tossico. Il deficit energetico o plastico viene compensato fornendo i metaboliti a valle dell’enzima che l’organismo non riesce più a produrre.
- Esempio: Nella Fenilchetonuria (PKU), si elimina la fenilalanina dalla dieta (compreso il dolcificante aspartame) integrando gli amminoacidi essenziali e la tirosina (che diventa un amminoacido essenziale per questi pazienti).
2. Sostituzione (Replacing)
Somministrazione diretta dell’ormone o del metabolita mancante a causa del blocco genico.
- Esempio 1: Fornitura di ormone tiroideo nell’ipotiroidismo congenito.
- Esempio 2: Nel deficit di 21-idrossilasi (che converte progesterone e idrossiprogesterone in cortisolo e aldosterone, accumulando androgeni che causano virilizzazione femminile), si somministrano cortisone, mineralcorticoidi e aldosterone. La virilizzazione fetale può essere prevenuta somministrando cortisone alla madre durante la gravidanza.
3. Deviazione Metabolica (Diversion)
Sfrutta la presenza di vie metaboliche secondarie normalmente silenti o scarsamente attive, stimolandole farmacologicamente per eliminare i metaboliti tossici accumulati a monte.
- Esempio: Nel deficit di ornitina transcarbamilasi (OTC), enzima del ciclo dell’urea, l’accumulo di carbammilfosfato viene convertito in ammoniaca (), sostanza altamente neurotossica che causa grave disabilità intellettiva. Somministrando sodio benzoato in grandi quantità, si forza il legame chimico con la glicina per formare ippurato, che viene escreto stabilmente con le urine. L’escrezione di ippurato stimola la sintesi de novo di glicina, la quale consuma e rimuove ulteriore ammoniaca dal circolo.
Terapia Farmacologica con Small Molecules
Le small molecules (piccole molecole) sono composti di sintesi chimica, cofattori o vitamine a basso peso molecolare (da centinaia a migliaia di Dalton), capaci di penetrare facilmente nelle cellule. Vengono utilizzate per modulare direttamente l’attività genica o proteica:
- Agenti read-through: Permettono al ribosoma di ignorare e saltare un codone di stop prematuro (mutazione nonsense) inserendo un amminoacido casuale tramite un tRNA non specifico. Questo consente il completamento della traduzione di una proteina intera, sebbene potenzialmente mutata in un singolo residuo.
- Esempio: L’antibiotico amminoglicoside Ataluren, utilizzato in trial clinici per il ripristino del canale CFTR in forme specifiche di Fibrosi Cistica dovute a stop codon.
- Correttori: Farmaci che si legano a proteine che presentano un ripiegamento alterato (folding errato), stabilizzandole e favorendone la corretta glicosilazione e il trasporto verso la loro localizzazione fisiologica (es. la membrana cellulare).
- Potenziatori: Molecole che aumentano la funzione residua di una proteina già correttamente posizionata nella sua sede cellulare (es. aumentando il tempo di apertura o la conduttanza di un canale ionico).
- Somministrazione di cofattori: Fornitura in grandi dosi di cofattori enzimatici per potenziare l’attività di enzimi mutati che presentano una ridotta affinità per essi (es. somministrazione di BH4 nella Fenilchetonuria).
Terapia a Livello Proteico (Sostituzione Enzimatica)
Rappresenta il 31% degli approcci terapeutici. Consiste nella produzione in vitro della proteina wild-type e nella sua successiva somministrazione al paziente (es. Fattore VIII ricombinante nell’Emofilia).
Limitazioni Storiche e Cliniche:
- Rischio patogeno: Storicamente, l’estrazione da donatori umani ha causato trasmissioni accidentali di patogeni (es. virus HIV in pazienti emofilici). Oggi si ricorre a proteine ricombinanti prodotte in colture cellulari.
- Reazione immunitaria: Trattandosi di proteine non-self (soprattutto in pazienti con totale assenza del gene), possono stimolare la produzione di anticorpi neutralizzanti.
- Emivita: Molte proteine vengono eliminate rapidamente dai reni. Per prolungarne la permanenza nel sangue, le si coniuga con il polietilenglicole (PEG) (es. nel trattamento del deficit di adenosina deaminasi, ADA).
- Barriere anatomiche: Difficoltà nel superare barriere come quella ematoencefalica per trattare manifestazioni neurologiche (es. nella malattia di Gaucher).
Modulazione dell’Espressione Genica
Consiste nel forzare o inibire l’espressione di specifici geni per compensare il difetto molecolare primario:
- Modulazione epigenetica (DNA): Uso di farmaci ipometilanti come la decitabina, che riducono la metilazione del DNA decondensando la cromatina e consentendo la riattivazione di geni spenti durante lo sviluppo (es. riattivazione dell’emoglobina fetale per compensare il deficit di nella Beta-Talassemia e nell’Anemia Falciforme).
- Exon skipping (RNA): Utilizzo di oligonucleotidi antisenso (ASO) che si legano a livello del pre-mRNA inducendo lo splicing a saltare l’esone mutato. Il mantenimento della griglia di lettura (frame) consente la produzione di una proteina più corta ma parzialmente funzionale (es. nella Distrofia Muscolare di Duchenne e Becker).
- Ormoni: Modulazione ormonale per aumentare l’espressione genica (es. uso di androgeni per indurre la sintesi dell’inibitore di C1-esterasi nell’angioedema ereditario).
⚠️ Indicazioni del docente
- Si raccomanda lo studio approfondito delle metodiche di diagnosi prenatale (amniocentesi e villocentesi), in quanto argomento frequentemente richiesto in sede d’esame.
🔗 Collegamenti
- Malattie Genetiche — 📋 fa parte di
- Terapia Genica — ⬇️ conseguenza
- Fibrosi Cistica — 💊 trattamento
- Fenilchetonuria — 💊 trattamento
- Ipercolesterolemia — 💊 trattamento
- Sindrome di Marfan — 💊 trattamento
- Distrofia Muscolare di Duchenne e Becker — 💊 trattamento
- Beta-Talassemia — 💊 trattamento
- Anemia Falciforme — 💊 trattamento
- Corea di Huntington — 💊 trattamento
- Gametogenesi — 🔬 reperto diagnostico