Corea di Huntington
La corea di Huntington (o malattia di Huntington) è una grave patologia neurodegenerativa progressiva del sistema nervoso centrale.
Eziopatogenesi e Trasmissione
La patologia è a trasmissione autosomica dominante e rappresenta un esempio nell’uomo di allele letale dominante:
- È localizzata su un cromosoma non sessuale (autosoma).
- È sufficiente la presenza di un singolo allele mutato per determinare lo sviluppo della malattia (eterozigosi). L’allele letale dominante in singola copia causa inevitabilmente la morte dell’individuo.
- Se un individuo eredita l’allele mutato, tipicamente almeno uno dei due genitori presenta manifestazioni della patologia (salvo casi di penetranza incompleta o nuove mutazioni de novo).
- Se un figlio non eredita l’allele mutato, la trasmissione della patologia si interrompe definitivamente in quel ramo familiare.
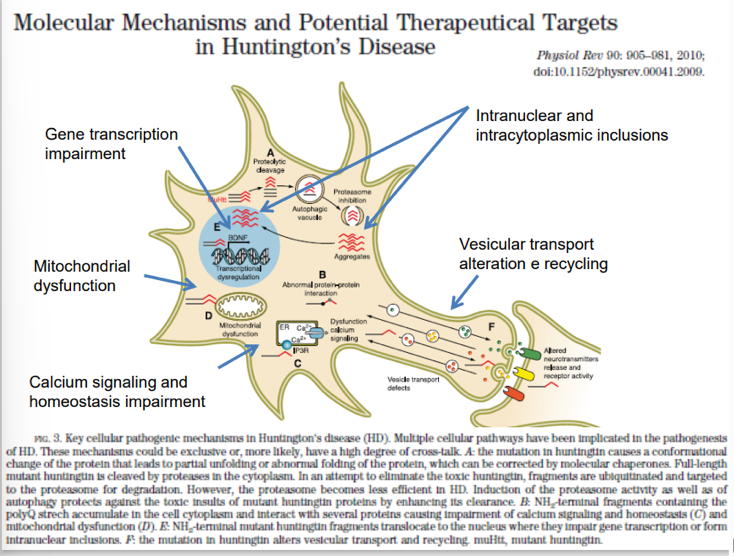
A livello molecolare, la malattia è associata all’errata sintesi e alla conseguente produzione in altissime quantità della proteina mutata huntingtina (causata dall’espansione di triplette nucleotidiche ripetute nel relativo gene).
(Integrazione da: sbobina 12)
- Meccanismo dell’Espansione delle Triplette: La mutazione consiste nell’espansione della tripletta ripetuta CAG, che codifica per la glutammina (creando una coda poliglutamminica patologica nell’huntingtina). L’instabilità di questa sequenza ripetuta è dovuta a una mutazione dinamica indotta da uno scivolamento replicativo (replication slippage) della DNA polimerasi. Durante la replicazione del DNA, la polimerasi si arresta, si distacca momentaneamente e si ri-appaia in modo scorretto scivolando indietro; questo determina l’inserimento di unità CAG addizionali nel filamento di neosintesi.
(Integrazione da: sbobina 13)
- Meccanismo Gain of Function: La Corea di Huntington rappresenta un modello di mutazione gain of function (acquisto di funzione) per acquisizione di nuove proprietà (novel properties). L’espansione CAG nel primo esone porta ad un allungamento anomalo della proteina (con una coda poliamminoacidica descritta come accumulo di glicine nella sbobina 13, sebbene a livello biologico CAG codifichi per la glutammina). Questo allungamento attrae e sequestra una serie di fattori trascrizionali a livello neuronale nel sistema nervoso centrale, determinando l’accumulo di aggregati tossici intracellulari e la degenerazione cellulare.
(Integrazione da: sbobina 28)
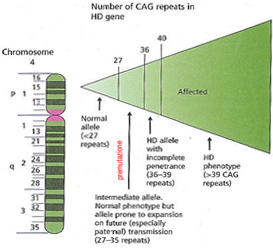
- Classificazione Allelica (Intervalli delle Triplette):
- Soggetti normali: fino a 26 ripetizioni (stabili, nessuna patologia).
- Premutazione / Zona grigia: ripetizioni. Gli individui non sono affetti ma la sequenza è instabile e a rischio di espansione nelle generazioni successive.
- Penetranza incompleta: ripetizioni. L’individuo potrebbe ammalarsi nel corso della vita o rimanere asintomatico.
- Affetti (mutazione completa): ripetizioni. Penetranza completa con espressioni cliniche inevitabili.
- Asimmetria Meiotica e Origine dell’Errore: L’instabilità dell’espansione differisce in base al sesso del genitore trasmettitore:
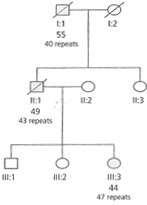
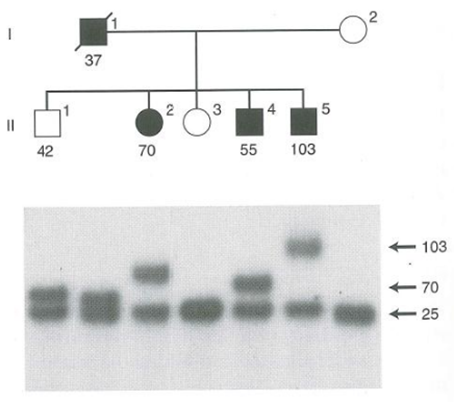
- Trasmissione Paterna (gametogenesi maschile): L’errore avviene durante la replicazione (mitosi dei precursori germinali) e non per ricombinazione meiotica. C’è un’elevatissima probabilità di ulteriore espansione del numero di triplette, portando a un esordio significativamente più precoce della malattia nella prole (anticipazione). Il padre affetto trasmette un rischio del 50% di generare figli affetti con espansione.
- Trasmissione Materna (gametogenesi femminile): La trasmissione è stabile. Una madre portatrice ha il 50% di probabilità di trasmettere la mutazione, ma il figlio non correrà il rischio di sviluppare un’espansione ulteriore o un fenotipo più grave (nessun aumento dell’anticipazione).
Quadro Clinico e Insorgenza
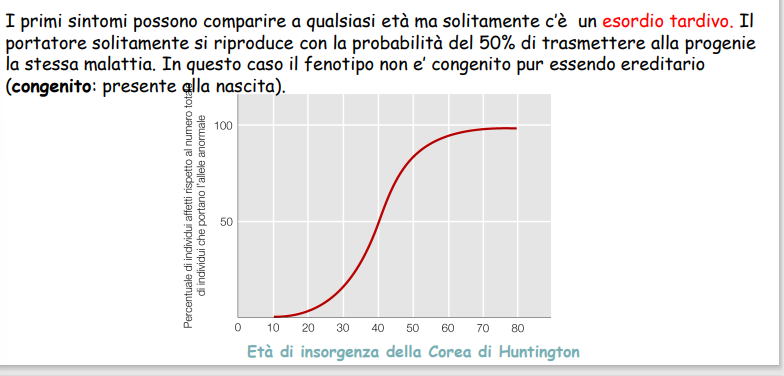
- Manifestazione Tardiva: I sintomi insorgono tipicamente in età adulta (spesso tra i 30 e i 50 anni, con un’età media intorno ai 40 anni). La penetranza è legata all’età: all’età di 50 anni, circa l’80% dei soggetti portatori della mutazione completa manifesta la sintomatologia clinica. L’insorgenza tardiva consente la riproduzione prima del decesso, permettendo la trasmissione alla prole.
- Sintomatologia Progressiva:
- Fase iniziale: Movimenti involontari e inappropriati delle estremità (mani e piedi), del collo e della testa, irrequietezza motoria ed iperreflessia.
- Fase avanzata: Deterioramento motorio grave (bradicinesia, distonia, rigidità muscolare) associato a progressivo declino cognitivo e demenza.
- Anticipazione Genica: Mostra il fenomeno dell’Anticipazione genica, manifestandosi a un’età più precoce e con sintomatologia progressivamente più grave nelle generazioni successive a causa dell’instabilità dell’espansione delle triplette nucleotidiche per via paterna.
- Segno Patognomonico Radiologico: L’unico segno specifico che supporta la diagnosi clinica in vivo è la presenza di atrofia del nucleo caudato, rilevabile tramite tomografia computerizzata (TAC) e risonanza magnetica nucleare (RMN).
(Integrazione da: sbobina 7)
(Integrazione da: sbobina 10) Rappresenta un modello fondamentale per lo studio della penetranza legata all’età (insorgenza tardiva) e dell’Anticipazione genica dovuta alla dinamica di espansione nucleotidica.
(Integrazione da: sbobina 16)
- Effetto del Fondatore: La malattia di Huntington rappresenta un esempio clinico classico di effetto del fondatore. In particolari comunità isolate geograficamente o socialmente (come le popolazioni nei pressi del Lago di Maracaibo in Venezuela o sull’isola di Tristan da Cunha), la presenza originaria di un singolo colonizzatore portatore dell’allele dominante patogeno ha determinato un’elevatissima frequenza della patologia nelle generazioni successive a causa del pool genico ristretto.

Prevenzione, Diagnosi e Gestione Terapeutica
Trattandosi di una malattia neurodegenerativa ad insorgenza tardiva e a trasmissione autosomica dominante:
- Approccio preventivo: La prevenzione (tramite counseling familiare e test genetico presintomatico nei soggetti a rischio) rappresenta l’intervento cardine. L’obiettivo delle strategie terapeutiche attuali e future è individuare precocemente i portatori dell’espansione nucleotidica prima della comparsa dei sintomi motori, per ritardare o prevenire l’esordio clinico della patologia.
(Integrazione da: sbobina 18)
(Integrazione da: sbobina 28)
- Diagnosi Molecolare di Elezione: Si utilizza la tecnica del Southern Blotting per determinare con precisione assoluta il numero di ripetizioni sul gene HTT, consentendo di tracciare le espansioni generazionali ed eventualmente prevedere l’età stimata di insorgenza nella prole.
- Counseling Genetico e Tipologie di Test:
- Test Diagnostico: Offerto a soggetti sintomatici per confermare la diagnosi clinica tramite l’analisi dell’espansione genica.
- Test Predittivo (Presintomatico): Offerto a soggetti asintomatici aventi un genitore affetto (rischio del 50%). Data la gravità prognostica dell’esito, il percorso deve essere condotto da un team multidisciplinare (genetisti, neurologi, psicologi) per preparare adeguatamente il paziente all’impatto psicologico e sociale della diagnosi.

🔗 Collegamenti
- Mutazione — ⬆️ causa
- Mutazione Spontanea — ⬆️ causa
- Mutazione Genica — ⬆️ causa
- Seconda Legge di Mendel — ⬆️ causa
- Genetica — 📋 fa parte di
- Alleli Letali — 📋 fa parte di
- Ereditarietà Autosomica Dominante — 📋 fa parte di
- Anticipazione — ⬆️ causa
- Classificazione Funzionale delle Mutazioni — 🔗 stesso meccanismo / stessa via
- Deriva Genetica — ⬆️ causa