Ipercolesterolemia
L’ipercolesterolemia familiare è una patologia genetica umana caratterizzata da elevati livelli di colesterolo lipoproteico a bassa densità (LDL) nel plasma, che determinano un elevato rischio cardiovascolare e coronarico precoce. Rappresenta un classico esempio clinico di dominanza incompleta (o parziale) nell’uomo.
Eziologia e Meccanismo Molecolare
La patologia è causata da mutazioni nel gene che codifica per il recettore delle LDL, una glicoproteina di membrana deputata al legame e all’internalizzazione delle particelle LDL circolanti nelle cellule (principalmente epatociti).
La sintesi dei recettori cellulari segue un modello a dosaggio genico dose-dipendente controllato da due alleli ( e ):
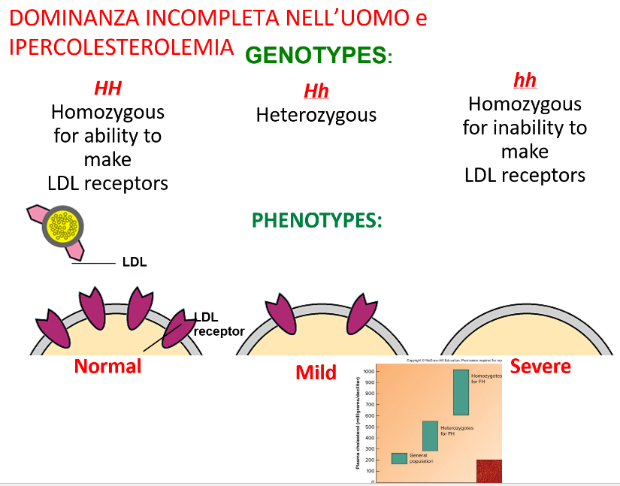
1. Genotipo Omozigote Dominante ( — Wild Type)
Gli individui omozigoti per l’allele funzionale sano riescono a produrre una quantità ottimale e completa di recettori LDL sulla membrana cellulare.
- Fenotipo: Sano. Le particelle LDL vengono efficacemente captate dal sangue e rimosse dal circolo per endocitosi.
2. Genotipo Eterozigote ( — Affetto Lieve)
Gli individui eterozigoti possiedono una sola copia dell’allele wild type e una copia dell’allele mutato non funzionale .
- Fenotipo: Sindrome clinica lieve o moderata. Le cellule producono circa la metà dei recettori LDL rispetto al soggetto sano, riducendo sensibilmente la capacità di captazione e internalizzazione. Questi soggetti presentano elevati livelli di colesterolo circolante sin dalla giovinezza e un rischio aumentato di infarto del miocardio e ostruzione arteriosa precoce.
3. Genotipo Omozigote Recessivo ( — Affetto Grave)
Gli individui omozigoti recessivi ereditano due copie dell’allele mutato e sono del tutto privi di recettori LDL funzionanti sulla membrana cellulare.
- Fenotipo: Patologia estremamente grave. Il colesterolo LDL non può essere internalizzato e rimane interamente nel torrente circolatorio, provocando aterosclerosi severa, placche occlusive e coronaropatie sin dall’infanzia.
Terapia
A differenza della popolazione generale sana (), i soggetti affetti richiedono un monitoraggio costante e un intervento terapeutico mirato:
- Eterozigoti (): Trattati principalmente con statine, farmaci inibitori dell’enzima HMG-CoA reduttasi, che bloccano la sintesi epatica endogena di colesterolo. Il conseguente deficit di colesterolo intracellulare induce come risposta compensatoria l’aumento dell’espressione genica dei recettori delle LDL; l’epatocita espone quindi un numero maggiore di recettori sulla propria superficie, catturando e rimuovendo efficacemente le LDL dal torrente circolatorio. Si associano modifiche dietetiche ed eventualmente inibitori di PCSK9.
- Omozigoti recessivi (): Essendo la superficie dell’epatocita completamente priva di recettori funzionanti, i geni dei recettori non possono essere sovraespressi in risposta alle statine. Non vi è quindi alternativa terapeutica all’aferesi periodica (un lavaggio e filtrazione meccanica extracorporea del sangue per rimuovere le LDL) o, nei casi più severi e refrattari, al trapianto di fegato.
(Integrazione da: sbobina 9)
- Nota di classificazione: Il docente segnala che l’ipercolesterolemia familiare viene da alcuni autori considerata una condizione codominante, sebbene questo aspetto non sia stato ulteriormente approfondito nel corso della lezione.
(Sezione espansa con: sbobina 18)
🔗 Collegamenti
- Estensioni delle Leggi di Mendel — ⬆ causa
- Allele — ⬆ causa
- Genotipo e Fenotipo — 🔬 reperto diagnostico
- Ereditarietà Autosomica Dominante — ⬆ causa