Beta-Talassemia
La -talassemia (-talassemia) è una forma di talassemia quantitativa caratterizzata da una ridotta o assente sintesi delle catene globiniche . È estremamente diffusa nel bacino del Mediterraneo; in Italia è particolarmente comune in Sardegna, Sicilia, Puglia, Calabria, e in specifiche aree del delta del Po e a Sant’Angelo Lodigiano.
Eziologia Molecolare ed Eterogeneità
A differenza dell’-talassemia (caratterizzata da delezioni), la -talassemia è causata prevalentemente da mutazioni puntiformi nel gene della -globina (cromosoma 11). Queste mutazioni colpiscono la trascrizione, lo splicing, la poliadenilazione o creano frameshift e codoni di stop prematuri.
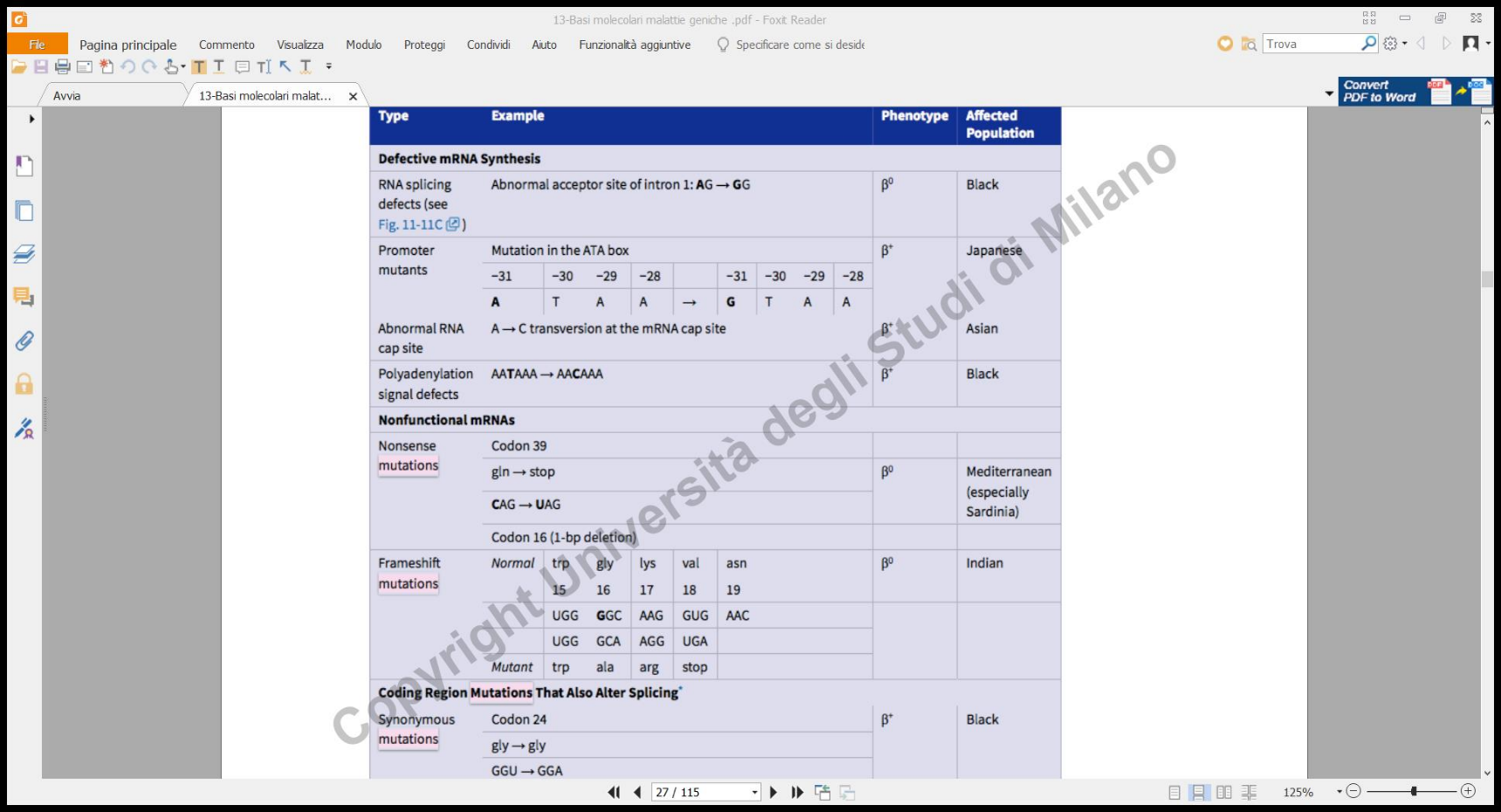
Classificazione Allelica
A seconda dell’impatto funzionale della mutazione, gli alleli sono classificati in:
- (Beta-zero): Totale assenza di produzione di catene funzionanti (mancanza completa dell’mRNA in omozigosi).
- (Beta-plus): Parziale riduzione quantitativa della sintesi di catene (forme talassemiche intermedie).
Le mutazioni mostrano una forte specificità geografica: ad esempio, la Sardegna presenta varianti specifiche dovute all’effetto del fondatore in una popolazione storicamente isolata e protetta dalla malaria.
Tratto -Talassemico (Eterozigote)
I soggetti eterozigoti ( o ) sono portatori sani asintomatici e presentano il cosiddetto tratto talassemico:
- Volume cellulare ridotto: Microcitosi costante (parametro diagnostico cardine).
- Anemia lieve: Livelli di emoglobina lievemente ridotti (~11 g/dL, rispetto ai fisiologici 13 g/dL).
- Trasmissione autosomica recessiva.

Talassemia Maggiore (Morbo di Cooley)
La forma omozigote o eterozigote composta (, ) determina una gravissima sindrome clinica nota come Morbo di Cooley.
Fisiopatologia e Decorso Clinico
- Insorgenza Postnatale: La sintomatologia non compare nel feto poiché in utero è espressa l’emoglobina fetale (). Si manifesta a partire dal 1°-2° anno di vita, in concomitanza con lo switch emoglobinico fisiologico verso l’HbA ().
- Danno Ossidativo ed Emolisi: La totale carenza di catene lascia libere le catene prodotte. I tetrameri di catene sono estremamente insolubili e precipitano all’interno degli eritroblasti e dei globuli rossi, provocando un danno ossidativo di membrana che innesca una massiva eritropoiesi inefficace a livello midollare ed emolisi splenica.
Manifestazioni Cliniche e Deformazioni Scheletriche
- Ipertrofia del Tessuto Ematopoietico: In risposta all’anemia e all’ipossia, il midollo osseo subisce un’espansione compensatoria massiva che deforma la struttura delle ossa (soprattutto quelle piatte del cranio, es. crani espansi, facies talassemica).
- Epatosplenomegaly: Milza e fegato si ingrossano notevolmente sia per far fronte alla distruzione dei globuli rossi alterati sia per la riattivazione dell’eritropoiesi extramidollare.
- Scarso Accrescimento: I bambini presentano deficit staturo-ponderale marcato dovuto all’anemia cronica.
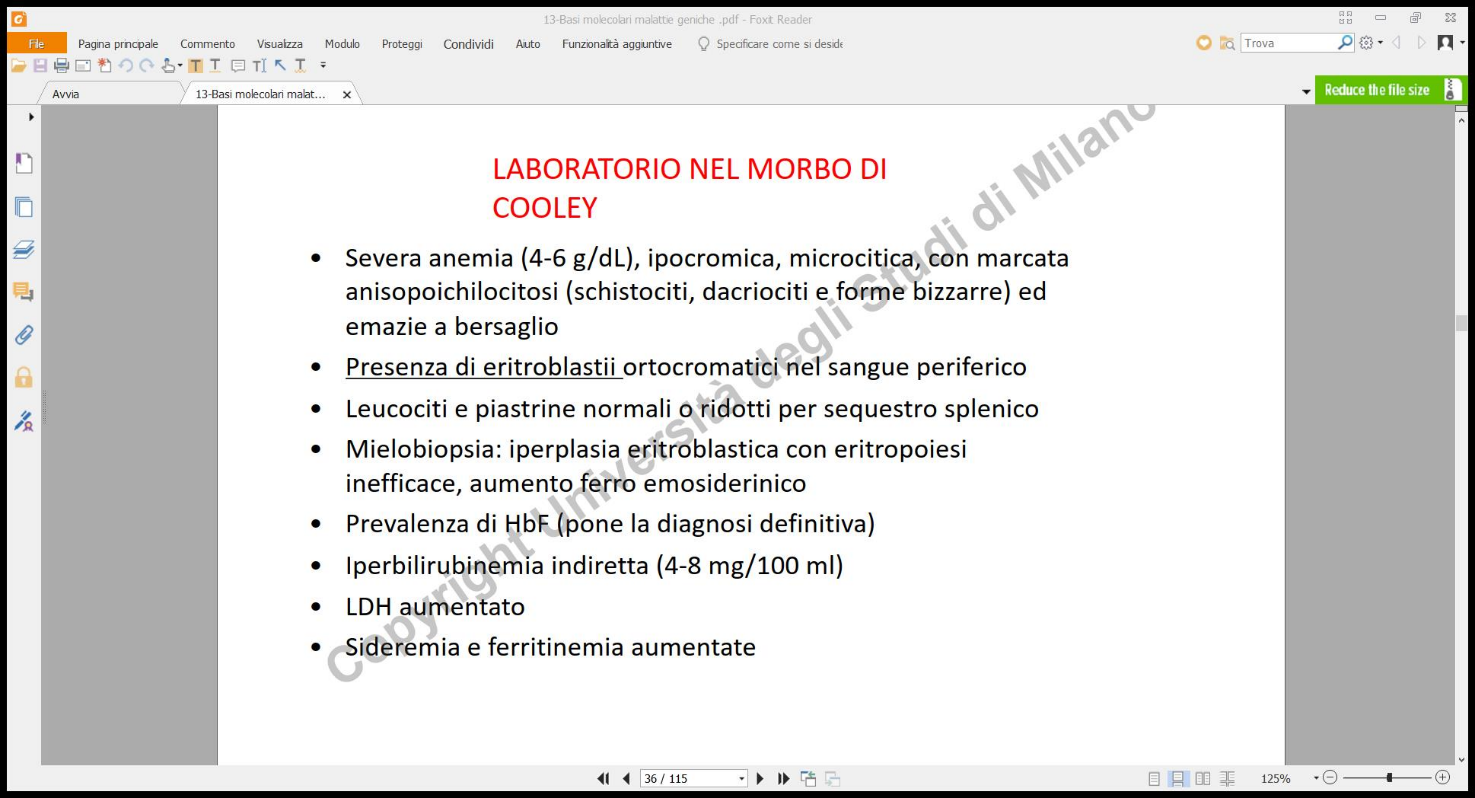
Reperti di Laboratorio
- Grave anemia ipocromica e microcitica (emoglobina molto bassa, globuli rossi piccoli e scarichi di colore).
- Anisopoichilocitosi: Presenza in circolo di globuli rossi con forme bizzarre e dimensioni estremamente disomogenee.
- Presenza di eritroblasti nucleati nel sangue periferico.
- Aumento della bilirubina indiretta (da emolisi), del ferro sierico e della ferritina.
Terapia e Complicanze da Sovraccarico (Emocromatosi Secondaria)
Il Morbo di Cooley richiede un approccio terapeutico cronico e multidisciplinare:
- Regime Trasfusionale: Trasfusioni ematiche costanti ogni 15 giorni circa per mantenere livelli di emoglobina vitali.
- Terapia Chelante del Ferro: Le continue trasfusioni causano la distruzione dei globuli rossi infusi e il conseguente rilascio massivo di ferro che l’organismo non può eliminare. Senza l’uso di chelanti del ferro, si sviluppa un’emocromatosi secondaria grave con deposizione tossica di ferro negli organi:
- Ipofisi: Causa deficit di ormone della crescita (GH) con arresto dello sviluppo e diabete insipido per deficit di ADH.
- Cuore: Cardiomiopatia siderotica, aritmie e insufficienza cardiaca.
- Pancreas: Deposizione nel pancreas endocrino con insorgenza di diabete mellito.
- Terapie Curative e Modulazioni Avanzate:
- Trapianto di midollo osseo: O di cellule staminali ematopoietiche sane (effettuabile anche in utero).
- Modulazione dell’espressione genica (Epigenetica): Somministrazione di decitabina, un agente ipometilante che decondensa la cromatina favorendo la riattivazione trascrizionale del gene per l’emoglobina fetale (-globina, Hb-γ). Le catene così prodotte si associano alle catene , sostituendo le catene mancanti e riducendo l’anemia e il danno da precipitazione di catene libere.
- Terapia genica (approccio ex vivo): Attualmente in fase di avanzata sperimentazione clinica (trial di fase I, II e III) con vettori lentivirali o adenovirali ricombinanti difettivi. Il protocollo prevede:
- Prelievo di midollo osseo autologo dal paziente affetto.
- Purificazione ed isolamento in vitro delle cellule staminali ematopoietiche esprimenti il marcatore CD34+.
- Trasfezione cellulare con il vettore virale contenente il cDNA del gene -globina wild-type.
- Selezione delle cellule staminali che mostrano una ottimale espressione del gene inserito, seguita da espansione clonale.
- Reinfusione (reimpianto) delle cellule staminali modificate nel paziente.
(Sezione espansa con: sbobina 18)
🔗 Collegamenti
- Talassemia — 📋 fa parte di
- Mutazione Genica — ⬆️ causa
- Emoglobina — 🔗 stesso meccanismo / stessa via