Anemia Falciforme
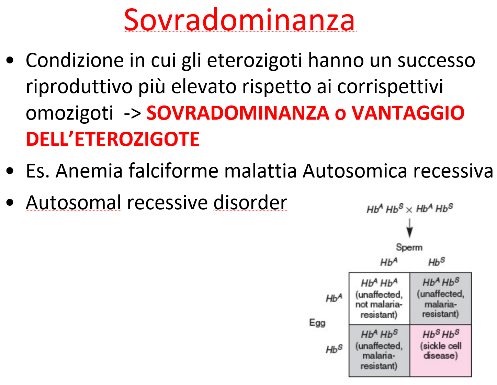
L’anemia falciforme è una patologia genetica autosomica recessiva caratterizzata da un’alterazione strutturale dei globuli rossi, che assumono una caratteristica forma a falce. Rappresenta un classico esempio clinico di sovradominanza (o vantaggio dell’eterozigote) in presenza del plasmodio della malaria.
Eziologia e Patogenesi Molecolare
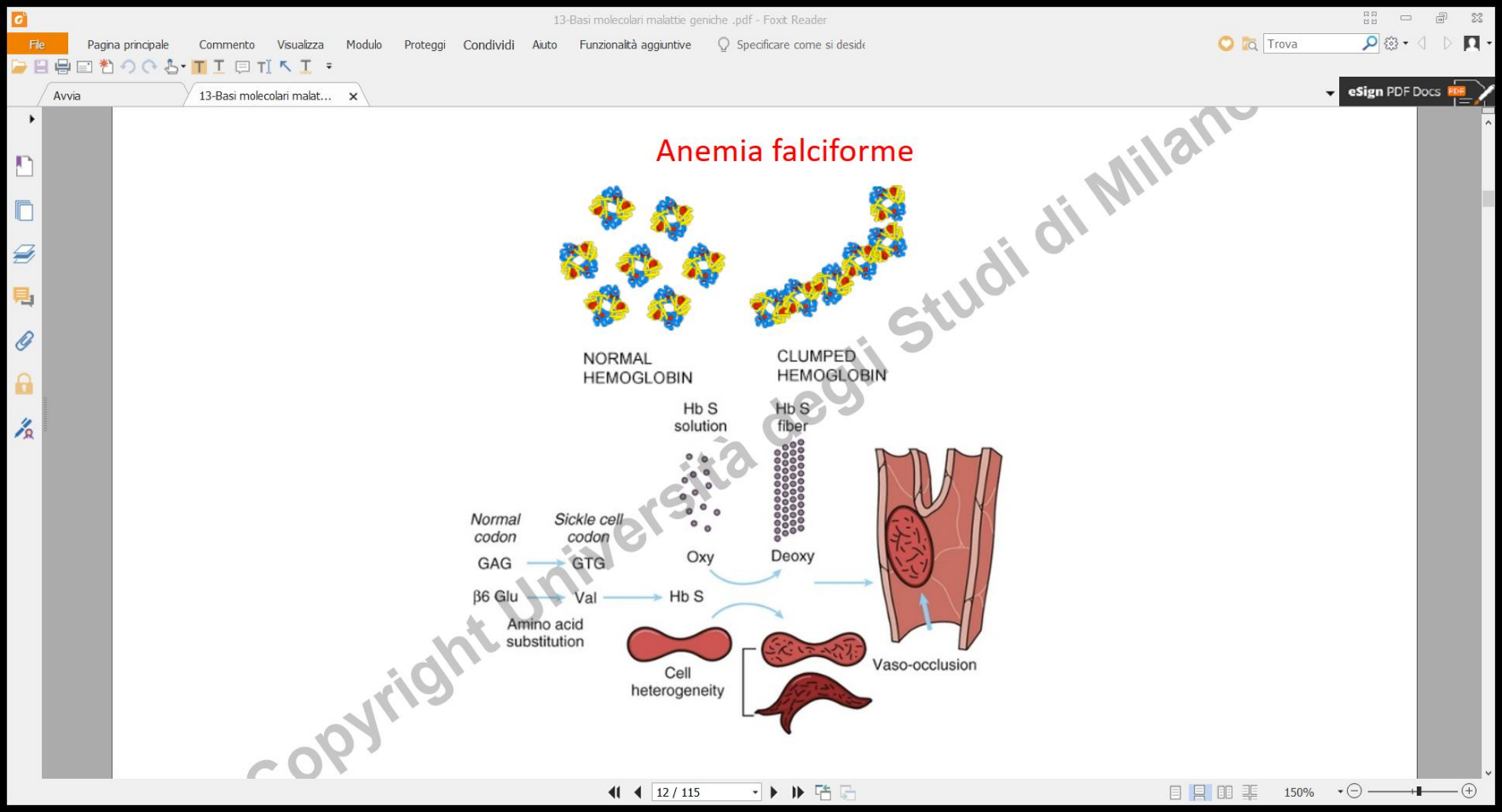
La malattia è causata da una mutazione missenso puntiforme nel gene che codifica per la catena dell’emoglobina (cromosoma 11). Tale mutazione sostituisce l’acido glutammico (polare e idrofilo) con la valina (apolare e idrofoba) in posizione 6.
La trasmissione è definita da due alleli:
- : Allele wild type (emoglobina normale).
- : Allele mutato (emoglobina falciforme).
Struttura e Comportamento dei Globuli Rossi
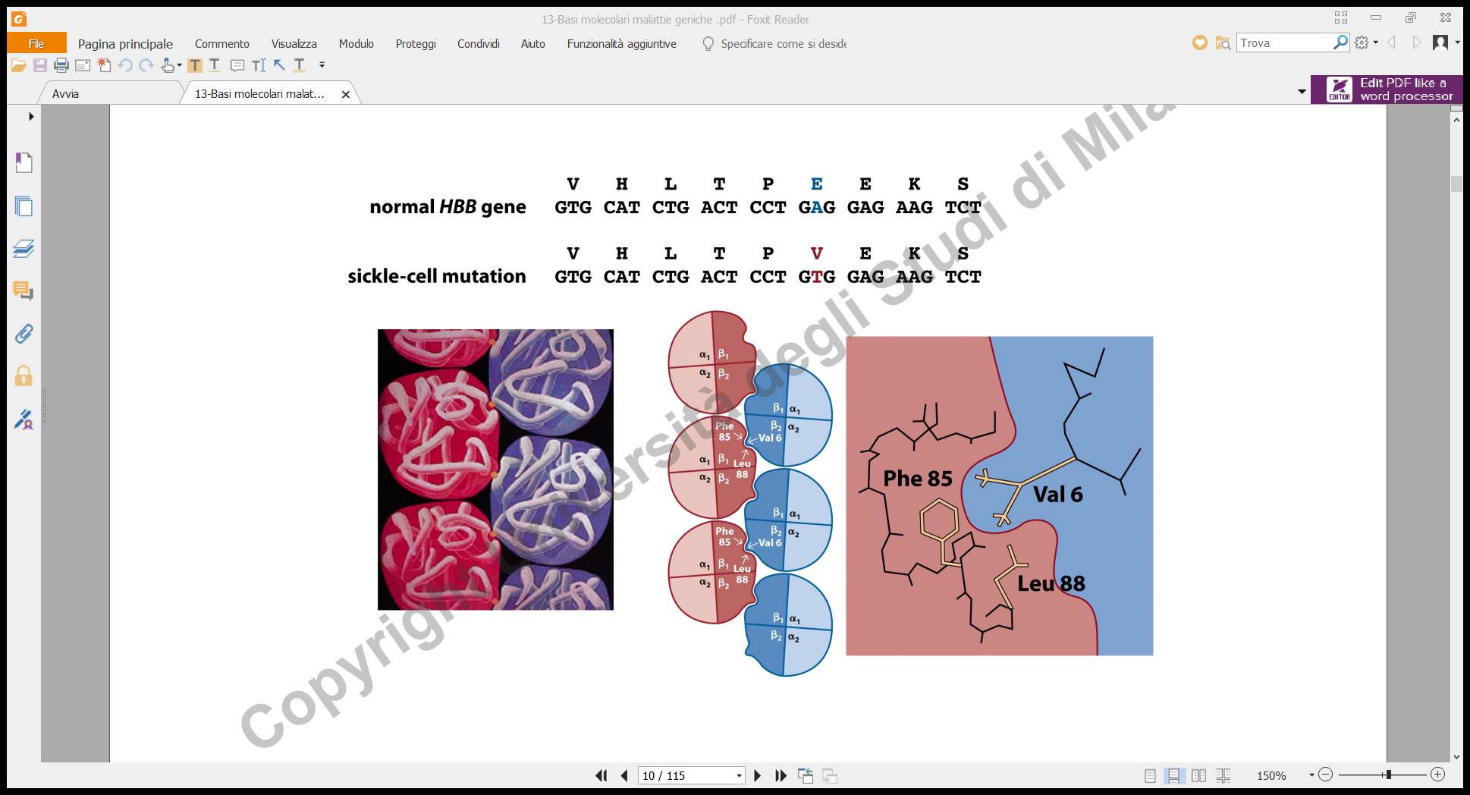
- Soggetti Omozigoti sani (): Producono emoglobina con struttura quaternaria corretta. I globuli rossi sono flessibili, biconcavi e scorrono facilmente nei vasi sanguigni.
- Soggetti Omozigoti affetti (): Producono esclusivamente emoglobina anomala. La presenza della valina (apolare) sulla superficie esterna della molecola deossigenata crea un patch idrofobico. In condizioni di bassa tensione di ossigeno (rilascio di ), la proteina subisce un cambiamento conformazionale e le molecole di emoglobina mutata polimerizzano tra loro formando lunghe fibre rigide e “appiccicose”. Queste fibre distorcono il globulo rosso conferendogli la rigida forma a falce. I globuli rossi falciformi vanno incontro a emolisi precoce (provocando grave anemia) e tendono a ostruire i capillari, causando crisi vaso-occlusive dolorose e danni ischemici d’organo.
- Soggetti Eterozigoti ( — Portatori sani): Possiedono un fenotipo clinico generalmente asintomatico o molto lieve. Solo una parte dell’emoglobina prodotta è difettosa, per cui i globuli rossi mantengono una stabilità sufficiente a evitare le crisi falciformi in condizioni fisiologiche normali.
(Integrazione da: sbobina 12)
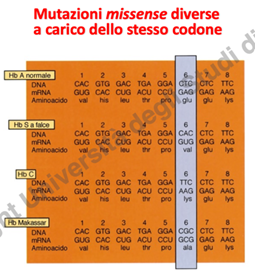
- Eterogeneità Allelica al Codone 6: La mutazione responsabile dell’anemia falciforme è una sostituzione missenso non conservativa del codone 6 del gene della -globina, che sostituisce l’acido glutammico (polare) con la valina (apolare). Il medesimo codone 6 rappresenta un hotspot mutazionale e può subire alterazioni differenti che portano a varianti qualitative distinte dell’emoglobina, come ad esempio:
- Emoglobina C (): Causata dalla sostituzione dell’acido glutammico con la lisina al codone 6.
- Altre varianti: Ulteriori sostituzioni alleliche qualitative al medesimo codone.
Sovradominanza e Resistenza alla Malaria
L’eterozigote , pur potendo mostrare lievi limitazioni funzionali in condizioni di ipossia estrema (es. ad altitudini elevate), possiede un enorme vantaggio evolutivo nelle aree geografiche dove la malaria è endemica.
Il Meccanismo del Vantaggio Eterozigote
Il ciclo vitale del Plasmodium falciparum (il parassita patogeno della malaria trasmesso dalla zanzara Anofele) richiede che il plasmodio permanga e si replichi all’interno del globulo rosso dell’ospite per circa 4 mesi.
- Nei soggetti sani , i globuli rossi hanno un’emivita regolare di 120 giorni, garantendo al parassita il tempo necessario per completare il ciclo infettivo e riprodursi, provocando la patologia malarica grave (spesso letale).
- Nei soggetti eterozigoti , la presenza dell’emoglobina mutata rende i globuli rossi più fragili. Se infettati dal plasmodio, questi globuli rossi vanno incontro a una senescenza e distruzione precoce (emolisi rapida).
- Poiché i globuli rossi infetti si spaccano prima che il plasmodio completi la maturazione, la carica parassitaria viene abbattuta e l’infezione viene interrotta sul nascere.
Significato Evolutivo
Sebbene la malaria sia stata debellata in molte regioni (es. bonifica delle paludi in Italia), la selezione naturale ha mantenuto una frequenza elevatissima dell’allele mutato nelle popolazioni originarie delle aree endemiche per la malaria (come l’Africa subsahariana). Nelle passate generazioni, la mortalità devastante causata dalla malaria ha favorito la sopravvivenza riproduttiva degli eterozigoti, consentendo la trasmissione della mutazione.
(Integrazione da: sbobina 8)
- Frequenza dei Portatori: In virtù del vantaggio evolutivo descritto, gli individui di origine africana (o provenienti da aree geografiche storicamente endemiche per la malaria) presentano una probabilità significativamente più elevata di essere portatori sani dell’allele .
(Sezione espansa con: sbobina 16)
Polimorfismo Bilanciato e Coefficienti di Selezione
Nelle aree in cui la malaria è endemica, si verifica una selezione differenziale che agisce contro entrambi gli omozigoti:
- I soggetti sani omozigoti ( o ) subiscono la pressione selettiva della malaria (mortalità elevata).
- I soggetti affetti omozigoti ( o ) muoiono precocemente per le crisi falciformi e la grave anemia.
- Gli eterozigoti ( o ), grazie alla parziale alterazione strutturale dell’emoglobina che impedisce la corretta replicazione del plasmodio, resistono alla malaria e non sviluppano la forma grave di anemia falciforme, avendo quindi una fitness massima ().
Questa selezione contrapposta contro entrambi gli omozigoti prende il nome di polimorfismo bilanciato. Essa mantiene stabili ed elevati entrambi gli alleli nella popolazione, in quanto la perdita dell’allele mutato negli omozigoti malati è compensata dalla perdita dell’allele sano negli omozigoti sani colpiti da malaria.
In assenza della malaria (es. grazie a interventi di bonifica), il coefficiente di selezione contro i soggetti omozigoti sani si azzera (, fitness ), eliminando il vantaggio degli eterozigoti e determinando una progressiva diminuzione della frequenza dell’allele mutato nelle generazioni future.
Effetti Pleiotropici e Quadro Clinico
L’anemia falciforme è una patologia pleiotropica classica. L’alterazione strutturale di un singolo gene (mutazione ) innesca una cascata di conseguenze patologiche che colpiscono organi e tessuti differenti:
- Lisi dei globuli rossi falciformi: Determina anemia grave che si manifesta clinicamente con astenia, debolezza costante e scarso sviluppo fisico.
- Crisi vaso-occlusive (occlusione dei capillari): La forma rigida a falce e la polimerizzazione delle fibre ostruiscono il microcircolo compromettendo l’irrorazione sanguigna. Questo flusso insufficiente genera ischemia, crisi di falcizzazione dolorose e danni d’organo a livello di:
- Cuore (cardiomiopatie ed insufficienza cardiaca)
- Cervello (paralisi e ictus)
- Polmoni (polmonite ed infarti polmonari)
- Tratto gastrointestinale (dolori addominali acuti)
- Reni (insufficienza renale da carenza d’ossigeno)
- Cute (ulcere cutanee croniche)
- Muscoli e articolazioni (reumatismo e alterazioni dolorose osteoarticolari)
- Accumulo splenico: Il sequestro e l’emolisi di grandi quantità di cellule falciformi nella milza ne provocano l’ingrossamento (splenomegalia) e la successiva fibrosi (fino all’autosplenectomia funzionale).
Geni Modificatori e Regolazione Clinica
La gravità clinica e la frequenza delle crisi dolorose variano tra i pazienti anche in base alla presenza di geni modificatori che modulano l’espressione dell’emoglobina fetale (-globina), la quale inibisce la polimerizzazione delle catene :
- Gene BCL11A: È un fattore di trascrizione che agisce fisiologicamente come repressore/silenziatore del gene . Polimorfismi nucleotidici presenti nell’introne tra l’esone 2 e 3 del gene BCL11A riducono la sintesi del repressore, riattivando la sintesi di catene e mitigando il fenotipo della malattia.
- Gene MYC (biologicamente MYB): Agisce anch’esso come inibitore della sintesi di . Polimorfismi nella regione del suo promotore (tra -84 e -77) ne riducono la produzione aumentandone i livelli di emoglobina F.
- Applicazioni terapeutiche: Questi meccanismi rappresentano la base molecolare per lo sviluppo di terapie geniche mirate a indurre farmacologicamente o geneticamente la riattivazione della sintesi di emoglobina fetale (HbF) nell’adulto.
(Integrazione da: sbobina 13)
(Integrazione da: sbobina 7)
Terapia
L’approccio terapeutico molecolare si avvale di strategie farmacologiche ed epigenetiche:
- Modulazione epigenetica (Decitabina): Somministrazione di decitabina, un farmaco ipometilante del DNA. Bloccando la metilazione del promotore del gene per la -globina (Hb-γ), la decitabina ne riattiva la trascrizione. L’emoglobina fetale (HbF, ) prodotta sostituisce funzionalmente le catene alterate ed impedisce efficacemente la polimerizzazione delle catene , prevenendo la falcizzazione del globulo rosso e riducendo le crisi vaso-occlusive dolorose.
- Terapia Genica: Sperimentazione di trial clinici avanzati per introdurre una copia corretta del gene -globina o indurre geneticamente il silenziamento del repressore BCL11A per promuovere la sintesi di HbF.
(Integrazione da: sbobina 18)
🔗 Collegamenti
- Mutazione — ⬆️ causa
- Mutazione Genica — ⬆️ causa
- Estensioni delle Leggi di Mendel — ⬆ causa
- Allele — ⬆ causa
- Seconda Legge di Mendel — 📋 fa parte di
- Genotipo e Fenotipo — 🔬 reperto diagnostico
- Pleiotropia — ⬆ causa
- Ereditarietà Autosomica Recessiva — 📋 fa parte di
- Classificazione Funzionale delle Mutazioni — 🔗 stesso meccanismo / stessa via
- Emoglobina — 📋 fa parte di
- Selezione Naturale — 🔗 stesso meccanismo / stessa via