Distrofia Muscolare di Duchenne e Becker
La distrofia muscolare di Duchenne (DMD) e la distrofia muscolare di Becker (BMD) sono miopatie progressive a trasmissione X-linked recessiva, determinate da alterazioni del gene della Distrofina. Rappresentano lo spettro clinico delle distrofinopatie.
Epidemiologia e Patogenesi
Le due patologie condividono lo stesso locus genico ma differiscono per il tipo di mutazione molecolare e per la gravità clinica:
- Distrofia Muscolare di Duchenne (DMD):
- Epidemiologia: Colpisce circa 1 su 3500 nati maschi.
- Meccanismo: Mutazioni out-of-frame (frameshift o stop codon prematuri) che causano l’assenza completa della proteina distrofina.
- Decorso: Insorgenza precoce nell’infanzia, progressione rapidissima, perdita della deambulazione autonoma e decesso tipicamente in età puberale/giovanile.
- Assenza di Manifestazione nelle Femmine (Sbobina 15): La DMD colpisce esclusivamente i maschi e non si osserva nelle femmine. Per manifestare la patologia (omozigosi recessiva), una femmina dovrebbe nascere dall’incrocio tra una madre portatrice ed un padre affetto. Tuttavia, poiché i maschi affetti muoiono generalmente prima di riprodursi (tra i 10 e i 15 anni), non possono trasmettere l’allele mutato alla progenie femminile.
- Distrofia Muscolare di Becker (BMD):
- Epidemiologia: Meno frequente, colpisce circa 1 su 18500 nati maschi.
- Meccanismo: Grandi delezioni in-frame che non alterano la griglia di lettura, consentendo la sintesi di una distrofina strutturalmente più corta ma parzialmente funzionale.
- Decorso: Insorgenza più tardiva, progressione lenta e interessamento muscolare meno severo.
Quadro Clinico e Sintomatologia
La sintomatologia clinica della DMD si manifesta precocemente e segue una progressione caratteristica:
- Esordio Motorio: Ritardo significativo nelle tappe motorie (il bambino cammina tardi trascinando i piedi). Compare difficoltà a correre, salire le scale, saltare e alzarsi da terra.
- Segno di Gowers: Per rialzarsi dalla posizione sdraiata o seduta sul pavimento, il bambino non riesce a fare forza sulle gambe a causa della debolezza dei muscoli prossimali del cingolo pelvico. Deve compiere una manovra caratteristica “arrampicandosi” su se stesso con l’ausilio delle mani e delle braccia.
- Anomalie Posturali e Deambulazione:
- Andatura anserina: Cammino oscillante simile a quello di una papera.
- Iperlordosi: Accentuazione della curvatura lombare per compensare la debolezza dei muscoli addominali e paravertebrali.
- Piede equino (cadente): Deambulazione sulle punte.
- Debolezza Distale ed Estensione: Con il progredire della patologia, la debolezza coinvolge gli arti superiori (difficoltà a scrivere, avvitare/svitare) e si manifestano contratture articolari.
- Pseudoipertrofia del Polpaccio: I polpacci appaiono volumetricamente aumentati; tuttavia, non si tratta di ipertrofia muscolare ma di sostituzione fibro-adiposa del tessuto muscolare necrotizzato.
- Coinvolgimento Sistemico:
- Cardiaco: Cardiomiopatia progressiva con aritmie. All’elettrocardiogramma (ECG) si osservano onde R prominenti nelle derivazioni precordiali destre e onde Q profonde nelle precordiali sinistre e periferiche.
- Gastrointestinale: Episodi di dilatazione gastrica acuta e disfagia (disturbi della deglutizione).
- Scheletrico: Demineralizzazione ossea e osteopenia indotte dal non uso e dall’immobilità.
- Respiratorio: Insufficienza respiratoria restrittiva a causa del coinvolgimento dei muscoli intercostali e del diaframma.



Iter Diagnostico
L’inquadramento diagnostico si articola su cinque livelli:
- Valutazione Clinica: Osservazione dell’andatura e riscontro del segno di Gowers.
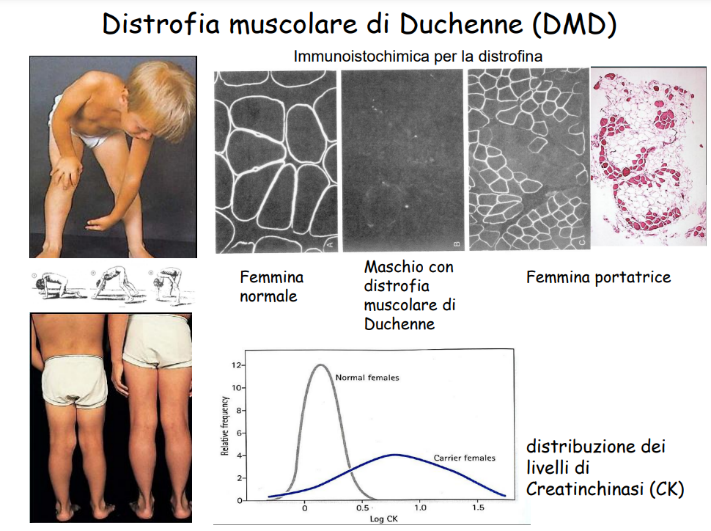
- Dosaggio della CPK (Creatina Fosfochinasi): Enzima muscolare rilasciato in circolo in seguito a necrosi della fibrocellula. Nei soggetti affetti da DMD, i livelli sierici di CPK risultano elevati fino a 100 volte rispetto alla norma.
- Elettromiografia (EMG): Esame neurofisiologico che valuta la risposta muscolare agli stimoli elettrici. Consente di escludere deficit primitivi di natura neurologica/neuropatica.
- Biopsia Muscolare: Prelievo bioptico per valutare l’alterazione istologica del tessuto (infiltrazione fibrotica) e per effettuare analisi immunoistochimiche sui livelli quantitativi di distrofina residua. Nota: è un esame invasivo che si tende a evitare se i test genetici sono dirimenti.
- Test Genetici:
- MLPA (Multiplex Ligation Probe Amplification) o CGH-Array: Consentono l’identificazione rapida di delezioni o duplicazioni esoniche (presenti nel 70-80% dei casi).
- NGS (Next Generation Sequencing): Utilizzato per sequenziare l’intero gene alla ricerca di microdelezioni o mutazioni puntiformi.

Gestione Terapeutica
L’approccio terapeutico molecolare moderno si focalizza su due principali fronti:
- Exon Skipping tramite ASO (Oligonucleotidi Antisenso): Nelle forme di Duchenne (DMD), caratterizzate da mutazioni out-of-frame che bloccano completamente la traduzione della Distrofina, si utilizzano gli ASO (anti-sense oligonucleotides). Queste molecole si legano al pre-mRNA inducendo l’apparato di splicing a saltare (skip) l’esone mutato, ripristinando il corretto frame di lettura. Sebbene si produca una proteina più corta (privata di un dominio non essenziale), essa risulta parzialmente funzionale. Questo converte clinicamente il fenotipo grave della Duchenne in un fenotipo molto più lieve e a progressione lenta, simile a quello della distrofia di Becker (BMD).
- Limitazioni della Terapia Genica Classica: L’utilizzo di vettori virali per inserire una copia sana del gene presenta due ostacoli principali:
- Dimensione del cDNA: Il gene della distrofina è estremamente grande e il relativo cDNA supera di gran lunga la capacità di carico dei comuni vettori virali (come gli AAV).
- Stato cellulare: I vettori virali infettano più efficacemente cellule in attiva divisione, mentre le cellule muscolari scheletriche (miociti) sono differenziate e post-mitotiche (non si dividono più), limitando fortemente la trasfezione e l’integrazione genica.
(Integrazione da: sbobina 18)
(Integrazione da: sbobina 27)
Manifestazione Clinica nelle Femmine Eterozigoti (Portatrici)
Sebbene la DMD sia classificata come una patologia X-linked recessiva che colpisce quasi esclusivamente i maschi, le femmine eterozigoti (portatrici) possono manifestare segni clinici a causa dell’Inattivazione del Cromosoma X.
- Ruolo della Lyonizzazione (Inattivazione dell’X): Nelle femmine, l’inattivazione casuale del cromosoma X determina un mosaicismo funzionale. Se in un tessuto muscolare o cardiaco la maggior parte delle cellule inattiva casualmente il cromosoma X sano (portando a un’inattivazione preferenziale o skewed dell’allele selvatico), le cellule esprimeranno solo la distrofina mutata.
- Variabilità Fenotipica: Questo meccanismo spiega perché le portatrici possano variare da una condizione del tutto asintomatica a forme di debolezza muscolare progressiva (mopatia simil-Duchenne/Becker) o a cardiomiopatia dilatativa isolata.
- Discordanza in Gemelle Monozigote: Nel caso di gemelle monozigote (geneticamente identiche), la scissione embrionale seguita da pattern indipendenti e asimmetrici di inattivazione del cromosoma X può determinare una discordanza fenotipica totale: una gemella può manifestare la distrofia muscolare (sebbene in forma più lieve rispetto al maschio emizigote) mentre l’altra gemella rimane una portatrice sana completamente asintomatica.
- Dosaggio della Creatinchinasi (CPK): Anche in assenza di sintomi motori evidenti, le femmine portatrici mostrano frequentemente livelli sierici elevati di creatinchinasi (CPK) a causa del danno subclinico continuo delle fibrocellule muscolari prive di distrofina funzionale.
🔗 Collegamenti
- Distrofina — ⬆ causa
- Distrofie Muscolari — 📋 fa parte di
- Ereditarietà Legata ai Cromosomi Sessuali — 📋 fa parte di
- Ereditarietà X-Linked Recessiva — 📋 fa parte di
- Eterogeneità Genetica — 🔗 stesso meccanismo / stessa via
- Mutazione — 🔗 stesso meccanismo / stessa via
- Inattivazione del Cromosoma X — ⬆ causa
- Gemelli — 🔄 diagnosi differenziale / confronto