Retinoblastoma
Il retinoblastoma è una neoplasia maligna oculare neuroepiteliale che colpisce quasi esclusivamente i bambini in tenera età. Rappresenta il modello classico per lo studio della genetica dei tumori basato sull’ipotesi dei due colpi di Knudson.
Eziologia Molecolare
La patologia è causata dalla perdita di funzionalità del gene RB (RB1, localizzato sul cromosoma 13q14), un oncosoppressore che codifica per la proteina pRb, regolatore cruciale del checkpoint di transizione tra la fase e la fase del Ciclo Cellulare. Trattandosi di una mutazione di tipo loss of function (perdita di funzione), la cellula necessita della perdita di entrambi gli alleli funzionanti per innescare lo sviluppo tumorale.
Forme Cliniche e Ipotesi dei Due Colpi (Knudson)
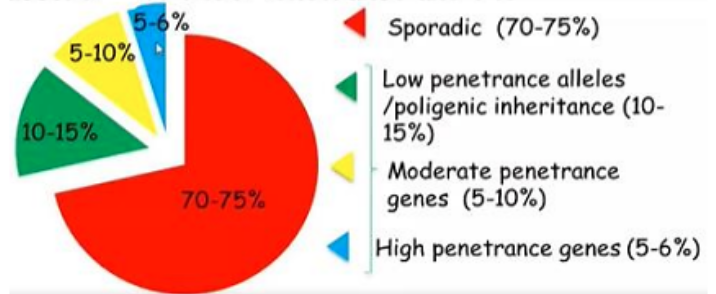
- Retinoblastoma Sporadico (70%):
- Gli individui nascono con due alleli wild-type integri.
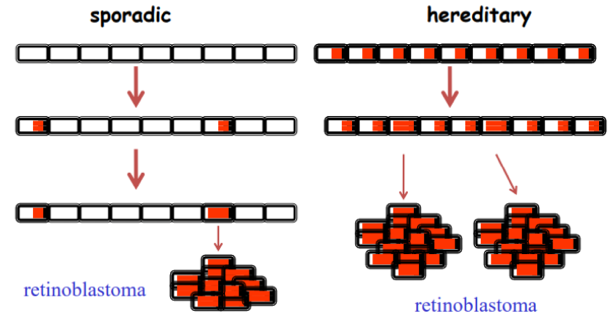
- Lo sviluppo del tumore richiede che avvengano due eventi mutazionali somatici indipendenti (due colpi) nella stessa cellula retinica.
- Di conseguenza, la forma sporadica si presenta tipicamente come un tumore unifocale (un solo tumore) e unilaterale (colpisce un solo occhio), con un’età d’insorgenza più tardiva rispetto alla forma ereditaria.
- Retinoblastoma Ereditario (30%):
- Gli individui ereditano costitutivamente un allele mutato per via germinale (primo colpo presente in tutte le cellule).
- Per l’insorgenza del tumore è sufficiente una singola mutazione somatica a carico dell’allele sano rimanente (second hit).
- La probabilità che questo secondo evento avvenga in almeno una delle cellule retiniche è elevatissima (comportamento clinico a trasmissione dominante ad alta penetranza).
- Questo si traduce in una presentazione clinica caratterizzata da tumori multifocali (più lesioni nello stesso occhio) e bilaterali (coinvolgimento di entrambi gli occhi), associata a un’età di insorgenza estremamente precoce.
Meccanismi del Second Hit
La perdita del secondo allele integro (perdita di eterozigosità o LOH) può verificarsi tramite mutazioni puntiformi, delezioni cromosomiche, o riarrangiamenti strutturali. Tuttavia, un meccanismo frequente di inattivazione dell’allele wild-type rimasto è rappresentato dal silenziamento epigenetico:
- Metilazione del Promotore: l’aggiunta di gruppi metilici a livello delle isole CpG nel promotore del gene RB ne blocca la trascrizione, determinando l’assenza della proteina funzionale pur in assenza di alterazioni fisiche della sequenza nucleotidica.
🔗 Collegamenti
- Oncogenesi — 📋 fa parte di
- Ciclo Cellulare — 🔗 stesso meccanismo
- Ereditarietà dei Tumori — 📋 fa parte di