Distrofia Miotonica di Tipo 1
La distrofia miotonica di tipo 1 (DM1), o malattia di Steinert, è una patologia multisistemica a trasmissione autosomica dominante caratterizzata da miotonia, progressivo indebolimento e atrofia muscolare, cataratta, difetti di conduzione cardiaca, alterazioni endocrine e cognitive.
Presenta una prevalenza stimata di circa 1 individuo affetto su 7500 nella popolazione.
Eziologia e Classificazione Allelica
La malattia è causata da una mutazione dinamica consistente nell’espansione di una ripetizione trinucleotidica CTG localizzata nella regione 3’ UTR (regione non tradotta al 3’) del gene DMPK1 (myotonic dystrophy protein kinase isoform 1), situato sul cromosoma 19.
A seconda del numero di ripetizioni CTG si distinguono diverse classi alleliche:
- Soggetti sani (normali): da 4 a 37 triplette.
- Soggetti affetti: da 50 a oltre 3000 triplette.
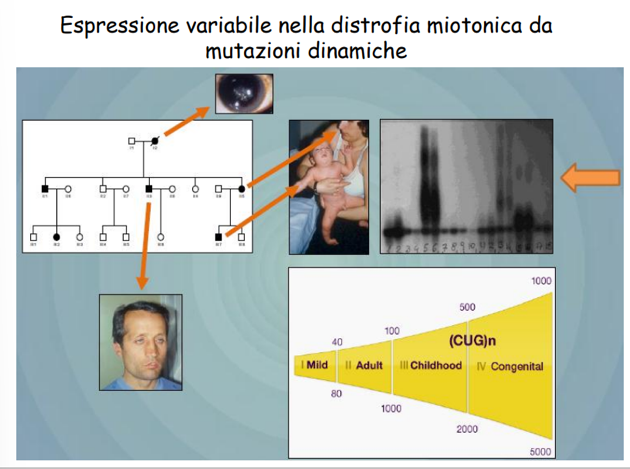
L’ampiezza dell’espansione correla strettamente con la gravità clinica e l’età di insorgenza (fenomeno dell’Anticipazione):
- Mild (lieve): ripetizioni. Caratterizzata prevalentemente da cataratta e miotonia lieve ad esordio tardivo.
- Classica: ripetizioni. Caratterizzata da miotonia severa, debolezza muscolare progressiva, cataratta e difetti di conduzione cardiaca.
- Congenita: ripetizioni. La forma più grave, caratterizzata da severa ipotonia neonatale, insufficienza respiratoria, difficoltà di apprendimento e rischio di mortalità precoce.

Ereditarietà e Influenza del Sesso Genitoriale
La DM1 mostra il fenomeno dell’anticipazione genica. L’instabilità del microsatellito e la sua tendenza all’espansione differiscono nettamente in base al sesso del genitore che trasmette la mutazione:
- Trasmissione Paterna (gametogenesi maschile): L’espansione durante la spermatogenesi è assente o di entità modesta. Di conseguenza, circa il 50% della prole eredita un quadro clinico e un’età di insorgenza simili a quelli del padre affetto.
- Trasmissione Materna (gametogenesi femminile): La meiosi femminile è fortemente instabile e predispone a espansioni massive. In caso di madre affetta o portatrice:
- Il 29% della prole manifesta una forma tardiva o classica simile a quella materna.
- Il 12% va incontro a morte neonatale.
- Il 9% manifesta la forma congenita (ipotonia neonatale e grave ritardo/difficoltà di apprendimento).

Esempio di Progressione Familiare
L’anticipazione genica e l’aggravamento progressivo dei sintomi tra le generazioni sono evidenti nei pedigree familiari:
- Generazione I: Soggetto affetto da cataratta (forma lieve).
- Generazione II: Figlio affetto da forma classica, con miotonia dei muscoli facciali, ptosi palpebrale (abbassamento di una o entrambe le palpebre) ed espressione facciale tipica.
- Generazione III: Nipote affetto da forma congenita ad insorgenza precocissima.

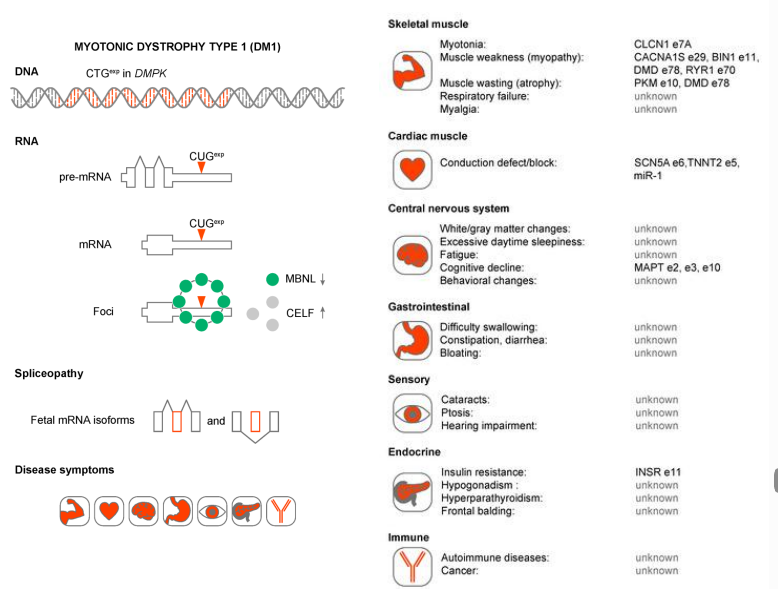
Patogenesi Molecolare: Gain of Function dell’mRNA
La DM1 non è causata da una perdita di funzione del gene DMPK1 (topi knockout per il gene non sviluppano il fenotipo della malattia), bensì da un meccanismo di acquisto di funzione (gain-of-function) dell’mRNA mutato:
- Trascrizione dell’espansione: La tripletta CTG sul DNA viene trascritta in CUG sul pre-mRNA di DMPK1.
- Sequestro delle proteine di splicing: Le triplette CUG espanse (fino a migliaia) assumono una conformazione secondaria stabile a forcina che recluta e sequestra massivamente proteine leganti l’RNA coinvolte nella regolazione dello splicing alternativo, in particolare le proteine CELF (CUGBP Elav-Like Family) e MBNL (Muscleblind-Like).
- Formazione di Foci Nucleari: I complessi formati dall’mRNA mutato e dalle proteine sequestrate precipitano nel nucleo cellulare, formando degli aggregati insolubili chiamati foci.
- Spliceopatia Multisistemica: La carenza di proteine CELF e MBNL libere impedisce il corretto splicing alternativo di pre-mRNA prodotti da altri geni non mutati. Questo difetto di splicing coordinato causa le diverse manifestazioni della patologia:
- Miotonia: Mancato splicing dell’RNA del gene CLCN1, che codifica per il canale principale del cloro del muscolo scheletrico (la perdita di funzione di CLCN1 causa direttamente la miotonia di Thomsen, dimostrando il nesso patogenetico).
- Difetti di conduzione cardiaca: Alterato splicing di specifici geni e microRNA espressi nel tessuto di conduzione cardiaca.
- Altri distretti: Lo splicing alterato coinvolge trascritti legati a funzioni endocrine, immunitarie e alla trasparenza del cristallino (cataratta).
🔗 Collegamenti
- Mutazioni Dinamiche — 📋 fa parte di
- Anticipazione — ⬆️ causa / fattore di rischio
- Talassemia — 🔄 diagnosi differenziale / confronto
- Distrofie Muscolari — 📋 fa parte di