Next-Generation Sequencing
Il Next-Generation Sequencing (NGS, sequenziamento di seconda generazione o sequenziamento massivo parallelo) comprende un insieme di tecnologie di sequenziamento degli acidi nucleici caratterizzate da un’altissima processività e velocità a costi estremamente ridotti rispetto al classico metodo Sanger.

IMPORTANT
Crollo dei costi di sequenziamento: Nel 2001, il completamento del Progetto Genoma Umano (e il sequenziamento del singolo genoma di Craig Venter) ha richiesto circa 100 milioni di dollari e anni di lavoro. Oggi, con le piattaforme NGS, un intero genoma umano può essere sequenziato in poche ore per un costo di circa 500$, consentendo l’analisi simultanea di decine di campioni (multiplexing).
Principali Approcci NGS
A seconda del quesito clinico e diagnostico, l’NGS viene impiegato secondo diverse modalità:

1. Whole Genome Sequencing (WGS)
Consiste nel sequenziamento dell’intero genoma dell’organismo (sia regioni codificanti che non codificanti).
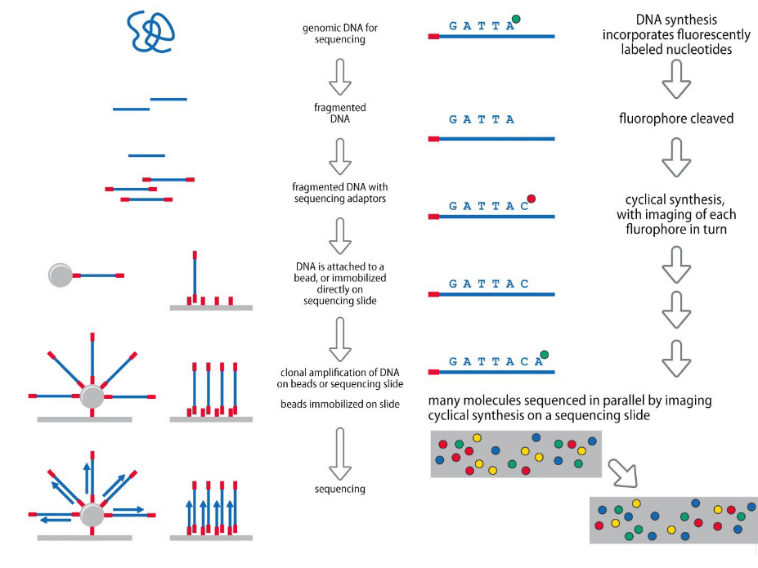
Flusso di lavoro:
- Estrazione e frammentazione: Il DNA genomico viene isolato e tagliato in frammenti di piccole dimensioni.
- Liaison degli adattatori e barcoding: Agli estremi dei frammenti vengono legati oligonucleotidi adattatori (necessari per l’ancoraggio e il sequenziamento) e sequenze specifiche di “codice a barre” (bar-code) che identificano il singolo paziente, consentendo di miscelare e sequenziare contemporaneamente fino a 90 campioni diversi.
- Amplificazione clonale: I frammenti vengono ancorati a un supporto solido e amplificati localmente (es. tramite PCR a ponte) per generare cluster clonali.
- Sequenziamento massivo parallelo: Sequenziamento in contemporanea di tutti i cluster.
- Allineamento: Allineamento bioinformatico delle letture (reads) ottenute rispetto a un genoma di riferimento. Limitazioni: Produce una quantità massiccia di dati complessi da gestire. Inoltre, le letture corte standard fanno fatica ad essere allineate in corrispondenza di sequenze ripetute. Per ovviare a questo problema, si utilizzano tecnologie di terza generazione (es. Nanopore) in grado di sequenziare singoli frammenti di DNA di grandi lunghezze.
2. Whole Exome Sequencing (WES)
Consente il sequenziamento selettivo del solo esoma, ossia dell’insieme degli esoni (regioni codificanti per proteine, che rappresentano circa l’1-2% del genoma ma contengono circa l’85% delle mutazioni patogenetiche note).
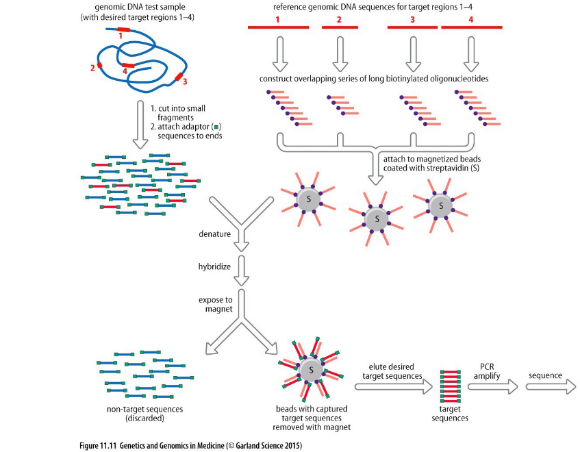
Flusso di lavoro (Cattura su Biglie Magnetiche):
- Il DNA genomico viene frammentato e addizionato di adattatori e barcode.
- Viene preparata una libreria di oligonucleotidi di cattura complementari a tutti gli esoni noti, coniugati con la biotina.
- Gli oligonucleotidi biotinilati vengono legati a biglie magnetiche rivestite di streptavidina (sfruttando l’altissima affinità del legame biotina-streptavidina).
- Le biglie magnetiche vengono incubate con il DNA frammentato del paziente: solo i frammenti di origine esonica si ibridano con le sonde sulle biglie.
- Utilizzando un magnete, le biglie (con gli esoni legati) vengono trattenute, mentre tutto il DNA non esonico (introni, regioni intergeniche) viene rimosso tramite lavaggio.
- Il DNA esonico viene eluito dalle biglie e sequenziato.
3. Esoma Clinico (Targeted Sequencing)
Consiste nel limitare il sequenziamento a un pannello selezionato di geni (es. 200-500 geni) già noti in clinica per essere associati a una determinata classe di patologie (es. cardiomiopatie, epilessie), riducendo ulteriormente costi e complessità interpretativa.
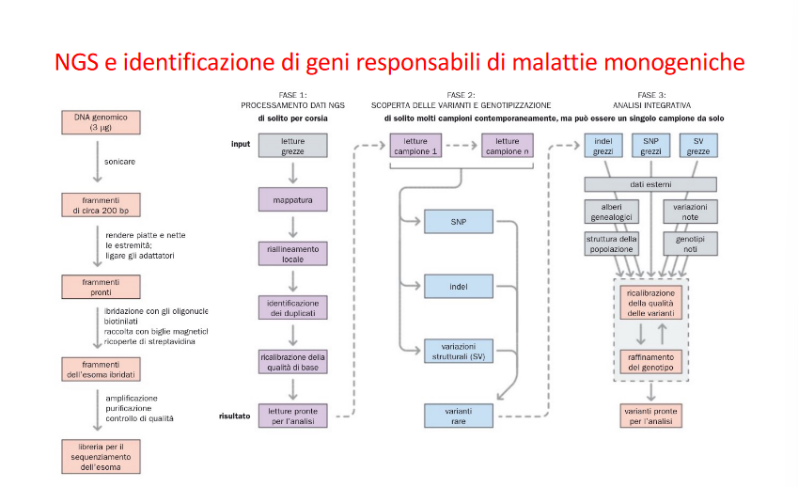
Workflow Bioinformatico di Filtraggio delle Varianti
Il sequenziamento del genoma o dell’esoma di un individuo rileva migliaia di varianti nucleotidiche rispetto al riferimento. Per individuare la mutazione causativa di una patologia, i dati vengono filtrati mediante un software dedicato:
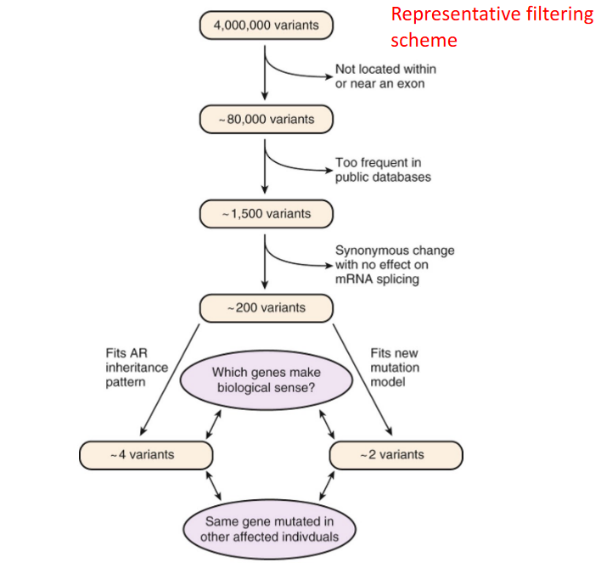
- Filtraggio di posizione: Rimozione di tutte le varianti localizzate in regioni non codificanti o non correlate all’ipotesi clinica (es. deserti genici).
- Filtraggio di frequenza nei database: Eliminazione delle varianti comunemente presenti nella popolazione generale (registrate in database come gnomAD). Poiché le malattie monogeniche gravi sono rare, la mutazione causativa deve presentare una frequenza allelica estremamente bassa o essere assente nei database.
- Predizione dell’impatto funzionale: Selezione delle varianti con elevata probabilità di danneggiare la proteina (mutazioni nonsense, frameshift, mutazioni dei siti di splicing, o missenso patogenetiche predette da algoritmi bioinformatici).
- Validazione diagnostica: Le varianti candidate residue (solitamente pochissime) vengono validate mediante sequenziamento classico Sanger sul paziente ed eventualmente sui genitori (analisi di segregazione familiare).
🔗 Collegamenti
- Medicina Personalizzata — 🔬 reperto diagnostico / strumento
- GWAS — ⬆️ causa / evoluzione
- Approccio del Gene Candidato — 🔬 reperto diagnostico / strumento
- Analisi di Linkage — ⬆️ causa / evoluzione