GWAS
Un GWAS (Genome-Wide Association Study, studio di associazione genome-wide) è un approccio di ricerca genetica utilizzato per associare variazioni genetiche — principalmente polimorfismi a singolo nucleotide (SNP) — a livello dell’intero genoma con una particolare malattia complessa o un tratto fenotipico.
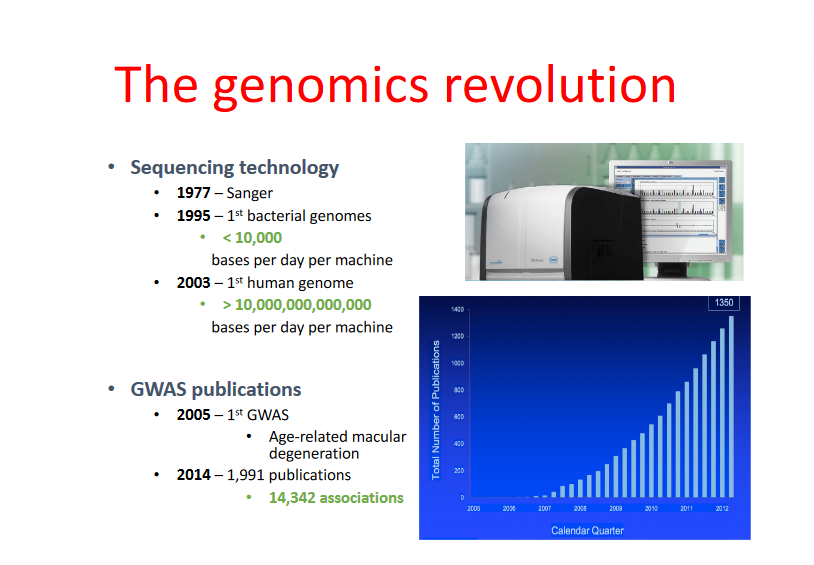
Metodologia e Flusso di Lavoro
L’analisi GWAS si basa su uno screening automatizzato ad alta processività condotto su ampie coorti di individui:
-
Selezione dei Marcatori: Si utilizzano gli SNP in virtù della loro elevata densità lungo il genoma (presenti in media ogni 1000-2000 coppie di basi).

-
Riduzione della Complessità (Tag-SNP): Sfruttando i dati del progetto HapMap sui blocchi di Linkage Disequilibrium, non si sequenziano tutti gli SNP del genoma, ma si seleziona un pannello di tag-SNPs rappresentativi di ciascun blocco aplotipico.

-
Genotipizzazione su Microarray: Il DNA di migliaia di casi e controlli viene ibridato su DNA chip (microarray) contenenti sonde specifiche per i tag-SNPs selezionati.
-
Analisi Statistica: Si confronta la frequenza di ciascun allele tra casi e controlli per individuare le varianti statisticamente associate alla malattia.
Rappresentazione dei Dati: Il Manhattan Plot
I risultati statistici di un GWAS vengono visualizzati graficamente tramite un Manhattan Plot:
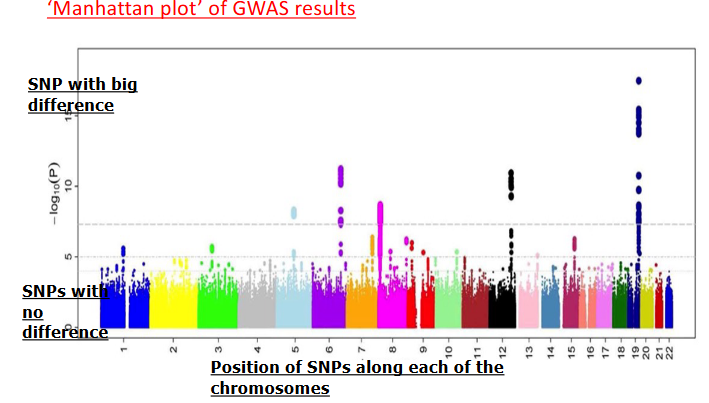
- Asse delle Ascisse (X): Posizione fisica delle varianti lungo i cromosomi (ordinati dal cromosoma 1 al cromosoma 22 e colorati alternativamente).
- Asse delle Ordinate (Y): Intensità dell’associazione statistica, espressa come il logaritmo negativo del p-value (). Più alto è il punto, maggiore è la significatività dell’associazione.
- Ogni punto rappresenta uno SNP testato. I segnali altamente significativi emergono dal rumore di fondo formando dei picchi verticali (“grattacieli”), indicanti la regione cromosomica contenente la variante associata alla malattia.
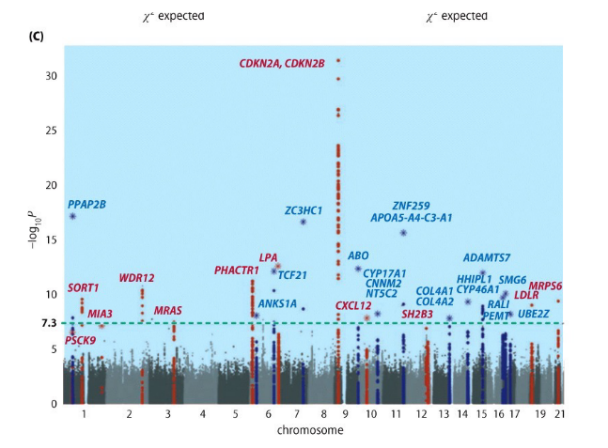
Esempio: Nel Manhattan plot sopra riportato, si osserva un picco prominente in corrispondenza del gene CDKN2A sul cromosoma 9, che ne dimostra l’associazione con il fenotipo studiato.
Limiti e Fallimenti del GWAS
Nonostante l’enorme mole di dati prodotta, l’approccio GWAS ha evidenziato diverse limitazioni nell’interpretazione dell’architettura genetica delle malattie complesse:

- Basso effetto delle varianti: La maggior parte delle associazioni individuate presenta un Odd Ratio molto basso; ciascun allele associato determina solitamente un incremento minimo del rischio relativo di malattia (es. circa il 5%).
- Ereditabilità Mancante (Missing Heritability): L’insieme dei segnali identificati tramite GWAS spiega solo una minima quota della componente genetica ereditaria complessiva stimata per le patologie multifattoriali.
- Deserti Genici: Molti picchi di associazione cadono in regioni intergeniche prive di geni noti (“deserti genici”), rendendo complessa l’identificazione del meccanismo biologico o del gene target regolato a distanza.
- Effetto Additivo Complesso: Poiché le malattie multifattoriali derivano dalla somma di moltissime varianti a basso effetto, risulta difficile mappare e comprendere l’interazione cooperativa di tutti i loci coinvolti.
Nello studio delle varianti rare a effetto moderato o alto, il GWAS viene integrato o sostituito dalle tecniche di sequenziamento massivo parallelo (Next-Generation Sequencing).
🔗 Collegamenti
- Studi di Associazione Genetica — 📋 fa parte di
- Aplotipo — 🔬 reperto diagnostico / strumento
- Next-Generation Sequencing — ➡️ evoluzione