Poliposi Adenomatosa Familiare
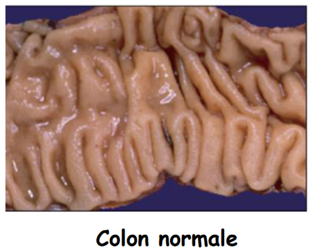
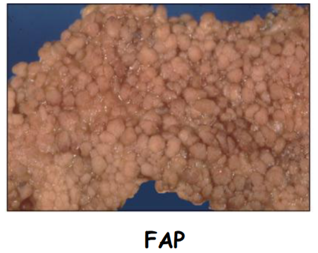
La Poliposi Adenomatosa Familiare (FAP) è una sindrome tumorale ereditaria a trasmissione autosomica dominante caratterizzata dalla comparsa di centinaia o migliaia di polipi (adenomi) lungo l’intero colon-retto. Se non trattata chirurgicamente mediante colectomia profilattica, l’evoluzione in adenocarcinoma colon-rettale presenta una probabilità del 100%.
Genetica Molecolare e Fisiopatologia
La FAP è causata da mutazioni loss of function (perdita di funzione) germinali a carico del gene APC (sul cromosoma 5q21-q22), un gene oncosoppressore.
- Ruolo di APC: codifica per una proteina che inibisce la via di segnalazione Wnt/beta-catenina, promuovendo indirettamente l’inibizione dell’espressione di MYC e di altri oncogeni. Partecipa inoltre ai meccanismi di adesione cellula-cellula (fondamentale per la corretta migrazione e differenziamento delle cellule della mucosa intestinale) e interagisce con la regolazione dei microtubuli durante la mitosi, controllando la corretta migrazione cromosomica.
- Penetranza e Correlazioni Genotipo-Fenotipo: la penetranza è elevata ma incompleta. Non essendovi hotspot mutazionali specifici in APC, si osservano quadri clinici variabili a seconda della posizione e della tipologia di mutazione:
- FAP Attenuata (AFAP): caratterizzata da un numero limitato di adenomi (solitamente meno di 100), insorgenza della patologia in età più tardiva e ridotta incidenza di manifestazioni extracoliche.
- Correlazione clinico-sintomatologica: specifiche mutazioni lungo il gene APC si associano, oltre alla predisposizione per i polipi intestinali, a un aumentato rischio di sviluppare manifestazioni extracoliche sistemiche quali:
- Ritardi della crescita
- Macrocefalia o microcefalia
- Cisti epidermoidi cutanee
- Tumori desmoidi (fibromatosi aggressive a livello della parete addominale o mesenterica)
- CHRPE (Ipertrofia congenita dell’epitelio pigmentato della retina)
MAP (Poliposi associata a MUTYH)
La MAP è una forma di poliposi adenomatosa clinicamente sovrapponibile alla FAP (con sviluppo di adenomi a livello del colon, dello stomaco e del duodeno) ma contraddistinta da un modello di ereditarietà autosomico recessivo.
- Eziologia: è causata da mutazioni a carico del gene MUTYH, il quale codifica per una glicosilasi coinvolta nei meccanismi di riparazione del DNA per escissione di basi (base excision repair), deputato a correggere i mismatch causati da stress ossidativo (es. 8-oxo-guanina).
- Le mutazioni in MUTYH presentano una penetranza variabile (bassa, media o alta) a seconda del tipo di variante allelica ereditata.
🔗 Collegamenti
- Tumori Ereditari del Colon-Retto — 📋 fa parte di
- Mutazione Genica — ⬆️ causa