Sindrome dell’X Fragile
La sindrome dell’X fragile (noto anche come sindrome di Martin-Bell) è una patologia genetica a trasmissione X-linked dominante con penetranza incompleta ed espressività variabile, caratterizzata da disabilità intellettiva e anomalie dello sviluppo.
Rappresenta la causa più comune di disabilità intellettiva di origine ereditaria nei soggetti di sesso maschile.

Cenni Storici ed Eziopatogenesi
- 1943: Martin e Bell descrivono la prima grande famiglia con maschi affetti da disabilità intellettiva.
- Identificazione del Sito Fragile: Analisi citogenetiche del cariotipo permisero di individuare un “sito fragile” (una costrizione secondaria) sul braccio lungo del cromosoma X (regione Xq27.3) indotta in condizioni di coltura specifiche (carenza di acido folico).
- 1991: Viene clonato il gene responsabile, FMR1 (Fragile X Messenger Ribonucleoprotein 1), rivelando che la fragilità citogenetica è dovuta all’espansione massiva di una ripetizione trinucleotidica CGG.
Meccanismo Molecolare e Classificazione Allelica
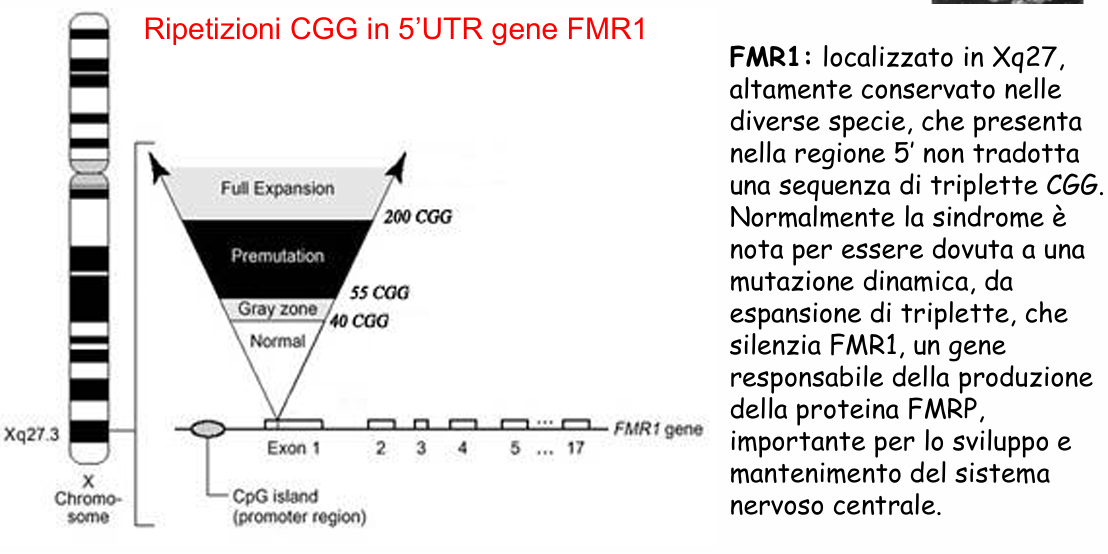
La mutazione si localizza nell’esone 1 (regione 5’ UTR non tradotta, parte del promotore) del gene FMR1. A seconda del numero di ripetizioni della tripletta CGG, si distinguono quattro classi alleliche:
- Assetto Normale: ripetizioni. La trasmissione ereditaria è stabile.
- Zona Grigia (Intermediate): ripetizioni. Gli alleli mostrano una lieve instabilità generazionale (aumento di ripetizioni) ma non causano patologia né espandono a mutazione completa nel passaggio successivo.
- Premutazione: ripetizioni.
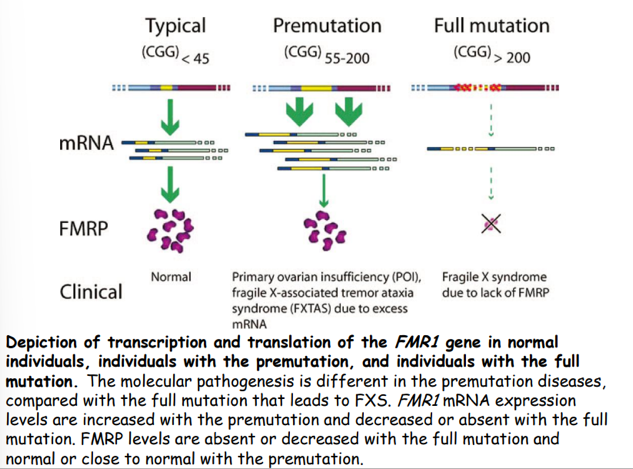
- Caratteristiche: Il gene è attivamente trascritto, ma l’allele è altamente instabile.
- Rischio di Espansione: La premutazione tende ad espandersi a mutazione completa durante la gametogenesi (meiosi) esclusivamente femminile. Il rischio di trasmettere la mutazione completa aumenta con la dimensione della premutazione materna (5% di rischio per ripetizioni, salendo al per ripetizioni). La trasmissione paterna della premutazione è stabile (non espande a mutazione completa).
- Mutazione Completa (Full Mutation): ripetizioni (fino a ).
- Silenziamento Epigenetico: L’espansione massiva oltre le 200 copie recluta metiltransferasi che inducono la metilazione delle isole CpG del promotore e la deacetilazione degli istoni, convertendo la cromatina in eterocromatina silente.
- Esito: Mancata trascrizione del gene FMR1 e totale assenza della proteina FMRP.
- Mutazioni Alternative: Circa il 2% dei pazienti presenta il fenotipo clinico classico della sindrome non a causa dell’espansione, ma per via di mutazioni puntiformi loss-of-function (es. delezione di basi o mutazioni non-senso) nel gene FMR1.
Il Paradosso di Sherman e l’Anticipazione
- Il Paradosso di Sherman: È il fenomeno per cui il rischio di manifestare la malattia aumenta nelle generazioni successive alla prima. Una madre apparentemente sana (portatrice di premutazione) ha un rischio elevato di avere figli maschi affetti (in cui la premutazione si è espansa a mutazione completa).
- Assenza di Anticipazione nella Mutazione Completa: A differenza di altre malattie da espansione di triplette (es. Corea di Huntington), nella Sindrome dell’X Fragile non si osserva anticipazione tra pazienti con mutazione completa. Poiché il gene viene silenziato al 100% una volta superate le 200 ripetizioni, un numero maggiore di triplette (es. 1000 o 2000) non aumenta la gravità dei sintomi né anticipa l’insorgenza della malattia.

Ruolo Fisiopatologico di FMRP e Neuropatologia
La proteina FMRP è una proteina legante l’RNA ampiamente espressa nel cervello e nei testicoli.
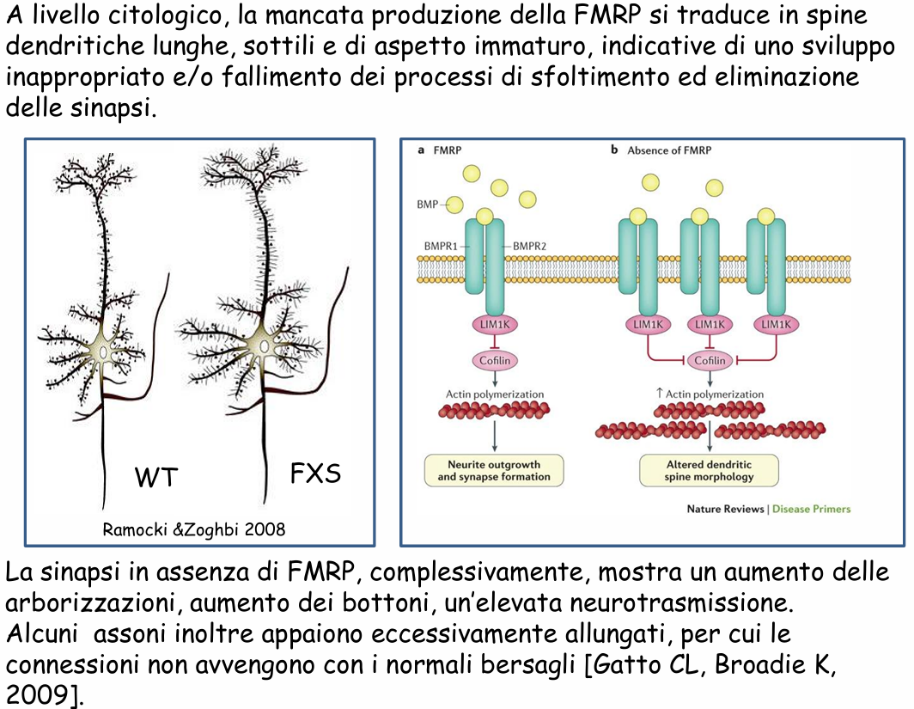
- Funzione cellulare: Regola il trasporto assonale e dendritico dell’mRNA, controlla la stabilità dei trascritti e funge da repressore della traduzione a livello locale nelle sinapsi eccitatorie (regolando la plasticità sinaptica a lungo termine). Partecipa inoltre alla morfogenesi dendritica e alla maturazione di astrociti e oligodendrociti.
- Effetto del Deficit (Morfologia Neuronale): L’assenza di FMRP determina un mancato controllo della traduzione sinaptica, provocando un’alterazione nella maturazione delle spine dendritiche: i neuroni degli affetti mostrano spine dendritiche eccessivamente sottili, lunghe, dense e iper-arborizzate, caratteristiche di uno stadio imaturo.
Quadro Clinico e Fenotipo
Il quadro clinico varia significativamente in base al sesso dell’individuo e all’età:
Soggetti di Sesso Maschile (Mutazione Completa)
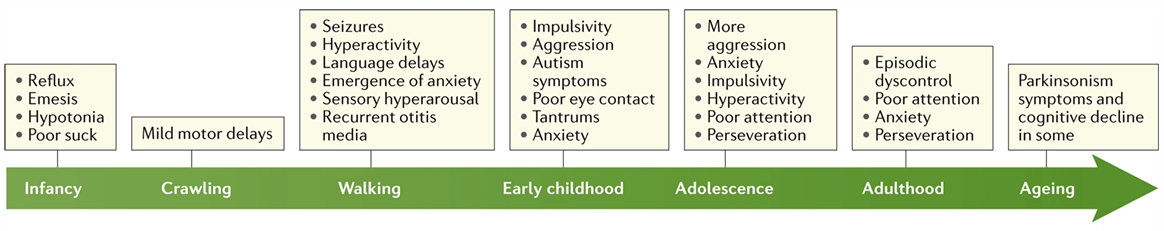
- Evoluzione del Fenotipo con l’Età:
- Età Infantile (sfumata): Difficoltà nella suzione, vomito frequente, ritardo motorio (gattonamento tardivo), iperattività, epilessia. Sono frequenti manifestazioni dello spettro autistico: ansia generalizzata, evitamento del contatto visivo prolungato, deficit dello sviluppo del linguaggio e stereotipie.
- Età Puberale/Adulta (peculiare): Sviluppo di dismorfismi facciali tipici quali viso allungato e grande, fronte prominente, ipoplasia zigomatica, padiglioni auricolari grandi e sporgenti. Si manifesta in modo caratteristico il macrorchidismo (volume testicolare nettamente superiore alla norma, visibile post-pubertà) e una diffusa lassità legamentosa.



Soggetti di Sesso Femminile (Mutazione Completa)
Presentano un fenotipo clinico significativamente più lieve e attenuato rispetto ai maschi. Questo è dovuto al fatto che, in eterozigosità, l’Inattivazione del Cromosoma X spegne casualmente il cromosoma mutato in circa il 50% delle cellule somatiche, che mantengono attiva la copia sana. Le femmine affette mostrano tipicamente:
- Ritardo cognitivo lieve o nei limiti della norma.
- Difficoltà specifiche di apprendimento.
- Tratti caratteriali di spiccata timidezza, introversione e ansia sociale.
Condizioni Associate alla Premutazione
I soggetti portatori di premutazione ( ripetizioni, con la sbobina 28 che evidenzia in particolare il range ) non sviluppano la sindrome dell’X fragile classica, ma sono fortemente predisposti a patologie specifiche dovute a un meccanismo di tossicità dell’mRNA di FMR1 (che viene trascritto in eccesso):
- Sindrome da Tremore e Atassia associata a X Fragile (FXTAS): Disturbo neurodegenerativo ad insorgenza tardiva caratterizzato da tremori intenzionali, atassia cerebellare e declino cognitivo.
- Insufficienza Ovarica Prematura associata a X Fragile (FXPOI): Disfunzione ovarica caratterizzata da menopausa precoce prima dei 40 anni, che colpisce il 20% delle donne portatrici della premutazione.
(Integrazione da: sbobina 28)
Inoltre, la trasmissione familiare delle premutazioni predispone ad un rischio elevato di espansione a mutazione completa nella prole (specialmente per via materna), manifestando quadri di FXS associati ad autismo, disordini dello spettro autistico (ASD) e ADHD.
Iter Diagnostico Molecolare
La diagnosi citogenetica basata sulla fragilità cromosomica è obsoleta per via della bassa sensibilità e specificità. Si utilizzano tecniche di genetica molecolare:
- Southern Blot: Metodica di riferimento storica. Permette di evidenziare sia lo stato di metilazione del promotore sia l’esatta dimensione dell’espansione genica (utile per distinguere la mutazione completa da premutazioni grandi).
- PCR PCR-Targeted (specifica per triplette): Metodica moderna che consente di identificare e quantificare con estrema precisione le ripetizioni CGG nei range normali, intermedi e di premutazione, ma che presenta limiti nella definizione precisa della taglia delle grandi espansioni (mutazione completa > 200), le quali faticano ad essere amplificate per l’elevato contenuto in GC.
🔗 Collegamenti
- Ereditarietà X-Linked Dominante — 📋 fa parte di
- Mutazioni Dinamiche — 📋 fa parte di
- Inattivazione del Cromosoma X — 🔄 diagnosi differenziale / confronto
- Anticipazione — 🔄 diagnosi differenziale / confronto
- Albero Genealogico — 🔬 reperto diagnostico
- Sindrome da Tremore e Atassia associata a X Fragile — ⬇️ conseguenza / complicanza
- Insufficienza Ovarica Prematura associata a X Fragile — ⬇️ conseguenza / complicanza