Sindrome di Beckwith-Wiedemann
La sindrome di Beckwith-Wiedemann (BWS) è una patologia da iperaccrescimento multisistemico ad eziologia epigenetica e genetica, con una prevalenza di circa 1 caso su 10500 nati vivi. I pazienti affetti mostrano alterazioni della crescita e dismorfismi caratteristici già alla nascita.
Quadro Clinico e Criteri Diagnostici (Consensus Score)
La diagnosi clinica della BWS si basa su un sistema di punteggio stabilito da linee guida internazionali (consensus), assegnando punteggi specifici alle diverse manifestazioni riscontrate:
Segni Cardinali (2 punti ciascuno):
- Macroglossia (lingua ingrossata).
- Esomfalo / Difetti della parete addominale (onfalocele, ernia ombelicale).
- Emi-ipertrofia / Lateralizzazione: Accrescimento corporeo asimmetrico (spesso causato da un mosaicismo epigenetico somatico).
- Tumori embrionali: Rischio marcatamente aumentato di sviluppare tumori nei primi anni di vita, come il tumore di Wilms (nefroblastoma), l’epatoblastoma, il neuroblastoma e il rabdomiosarcoma.
Segni Suggestivi (1 punto ciascuno):
- Pieghe auricolari posteriori tipiche o fossette lobi auricolari.
- Ipoglicemia neonatale.
- Nefromegalia o visceromegalia.
La diagnosi è posta al raggiungimento di un punteggio clinico minimo stabilito.

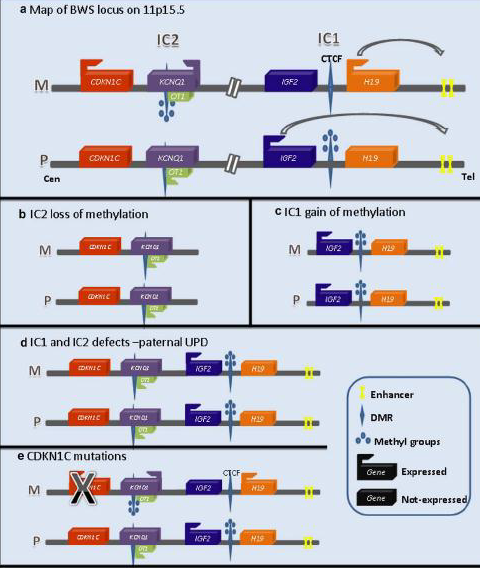
Eziopatogenesi Molecolare (Locus 11p15.5)
La BWS è causata da un’alterazione nel dosaggio trascrizionale di geni sottoposti ad imprinting sul cromosoma 11p15.5, in particolare dei fattori stimolanti la crescita (IGF2) e degli inibitori di crescita (CDKN1C).
Si riconoscono quattro meccanismi principali alla base della sindrome:
- Ipometilazione di IC2 sull’allele materno (60% dei casi):
- Meccanismo: La perdita patologica di metilazione del centro dell’imprinting IC2 ( KvDMR1) sull’allele materno attiva la trascrizione del lncRNA antisenso Lit1 (Kcnq1ot1). Lit1 promuove il silenziamento del gene CDKN1C (inibitore del ciclo cellulare) anche sull’allele materno. La perdita di questo “freno” cellulare causa l’iperaccrescimento.
- Ipermetilazione di IC1 sull’allele materno (10% dei casi):
- Meccanismo: L’acquisizione anomala di metilazione su IC1 sull’allele materno impedisce il legame dell’insulator CTCF. Di conseguenza, il gene H19 viene spento e gli enhancer attivano la trascrizione di IGF2 anche sull’allele materno. L’individuo presenta un doppio dosaggio di IGF2 (stimolatore di crescita).
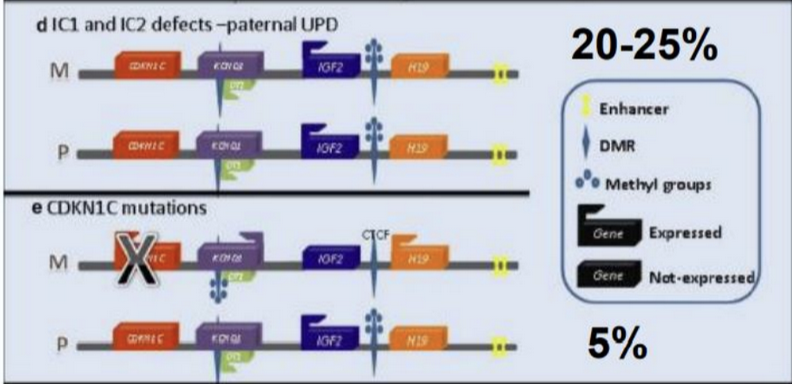
- Disomia Uniparentale Paterna - UPDpat (20 - 25% dei casi):
- Meccanismo: Ereditarietà a mosaico di due copie del cromosoma 11p15.5 di origine paterna (UPD). Questo causa simultaneamente l’ipermetilazione di IC1 (doppio dosaggio di IGF2) e l’ipometilazione di IC2 (assenza di CDKN1C).
- Rischio oncologico: I pazienti con UPDpat hanno il rischio più elevato di sviluppare tumori embrionali.
- Mutazione del gene CDKN1C (5% dei casi):
- Meccanismo: Mutazione puntiforme loss-of-function dell’allele materno di CDKN1C, che annulla l’attività dell’inibitore.
Calcolo del Rischio di Ricorrenza e Diagnosi di Laboratorio
La diagnosi molecolare viene effettuata analizzando lo stato di metilazione delle regioni IC1 e IC2. La perdita della metilazione differenziale fisiologica (ovvero se entrambi gli alleli appaiono metilati o entrambi non metilati) conferma la diagnosi.
L’identificazione del meccanismo molecolare esatto è indispensabile per calcolare il rischio di ricorrenza in gravidanze successive:
- Difetti Sporadici (Ipometilazione IC2, Ipermetilazione IC1, UPDpat): Il rischio di ricorrenza è basso, inferiore all’1% rispetto alla popolazione generale.
- Difetti Genetici Ereditabili (Mutazione di CDKN1C materna, duplicazioni o traslocazioni bilanciate della regione paterna): Il rischio di ricorrenza è elevato, pari al 50%.
🔗 Collegamenti
- Imprinting Genomico — ⬆️ causa
- Meccanismo dell’Imprinting — 📋 fa parte di
- Teoria del Conflitto Parentale — 📋 fa parte di
- Sindrome di Silver-Russell — 🔄 diagnosi differenziale / confronto
- Bisomia Uniparentale — ⬆️ causa
- Mosaicismo — ⬆️ causa