Neoplasie Endocrine Multiple
Le Neoplasie Endocrine Multiple (MEN) costituiscono un gruppo di sindromi tumorali ereditarie caratterizzate dallo sviluppo di lesioni neoplastiche iperplastiche o tumorali a carico di molteplici ghiandole e tessuti del sistema endocrino. L’inquadramento clinico e genetico si basa sul tipo specifico di MEN e sul coinvolgimento di differenti classi geniche (oncogeni e oncosoppressori):
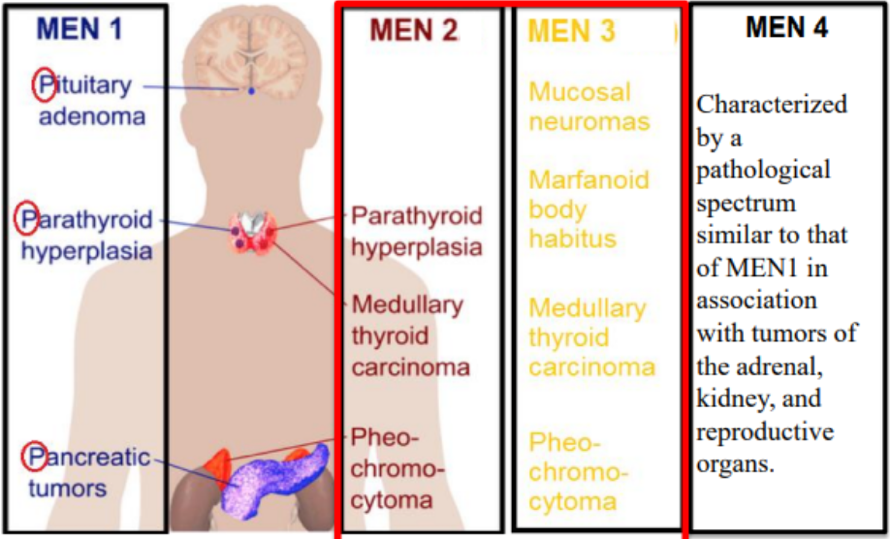
Classificazione Genetica delle MEN
MEN 2 e MEN 3 (MEN 2B)
Entrambe le varianti sono dovute a mutazioni del gene RET (localizzato sul cromosoma 10), un protooncogene che codifica per un recettore a tirosina chinasi.
- Meccanismo Patogenetico: le mutazioni sono di tipo gain of function (guadagno di funzione) e si localizzano in specifici domini strutturali del gene RET.
- Correlazione Genotipo-Fenotipo: la specifica localizzazione della mutazione all’interno dei domini del gene determina la manifestazione di MEN 2 o MEN 3, determinando quadri sintomatologici differenti.
- Espressione Clinica: l’ereditabilità si comporta in modo dominante. Tuttavia, la presenza della mutazione conferisce una predisposizione e non garantisce che ogni paziente esprima l’intero spettro di neoplasie tipiche della sindrome.
MEN 1
È determinata da mutazioni a carico del gene MEN1 (localizzato sul cromosoma 11), il quale si comporta come un classico gene oncosoppressore.
- Meccanismo Patogenetico: le mutazioni ereditate sono di tipo loss of function (perdita di funzione). Gli individui predisposti ereditano costitutivamente un singolo allele mutato (eterozigosi).
- Ipotesi dei Due Colpi (Knudson): trattandosi di un meccanismo recessivo a livello cellulare, la formazione di neoplasie si verifica esclusivamente a seguito della perdita o inattivazione del secondo allele normale (second hit o perdita di eterozigosità - LOH) in una cellula somatica.
Caso Clinico e Dimostrazione Molecolare del Second Hit
Un caso clinico documenta una paziente con diagnosi clinica certa di MEN 1 in cui la mutazione costitutiva non era inizialmente identificabile con le analisi molecolari standard. La paziente presentava:
- Iperparatiroidismo
- Iperplasia della paratiroide
- Macroadenoma ipofisario secernente prolattina
- Lesioni pancreatiche successivamente evolute in adenocarcinoma pancreatico
- Lipomi multipli localizzati agli arti inferiori
L’applicazione della tecnica di rCGH (Array-CGH su vetrino) ha permesso di chiarire il meccanismo molecolare nei diversi tessuti:
- Mutazione Costitutiva (Primo Colpo): l’analisi rCGH ha evidenziato una microdelezione costitutiva della regione contenente il gene MEN1 (cromosoma 11q13), presente in tutte le cellule della paziente.
- Adenoma Ipofisario (Second Hit): l’analisi dell’adenoma ipofisario ha rivelato la presenza della delezione in omozigosi del gene MEN1. Questo fenomeno è avvenuto a causa della perdita del cromosoma 11 integro e della concomitante duplicazione del cromosoma 11 contenente la delezione costitutiva, determinando la perdita completa di funzionalità dell’oncosoppressore nel tessuto tumorale.
- Tessuto del Lipoma (Second Hit): l’analisi citogenetica del lipoma ha individuato una traslocazione tra il cromosoma 3 e il cromosoma 11. Questa traslocazione ha determinato la rottura e la perdita della regione critica contenente il secondo allele integro di MEN1, determinando anche in questo tessuto il second hit necessario all’insorgenza neoplastica.
🔗 Collegamenti
- Oncogenesi — 📋 fa parte di
- Delezione Cromosomica — ⬆️ causa
- Traslocazione Reciproca — ⬆️ causa
- Array-CGH — 🔬 reperto diagnostico