Array-CGH
L’array-CGH (Array - Comparative Genomic Hybridization) è una tecnica di Citogenetica Molecolare che analizza l’intero genoma per identificare variazioni nel numero di copie di determinate regioni. Ha una risoluzione di poche kb (20-100 kb).
Phenotype first vs genotype first
- Phenotype first: si osserva il fenotipo del paziente, lo si associa a una condizione patologica nota e si conferma il sospetto clinico con la FISH, usando sonde complementari alla regione da indagare. Il fenotipo riconduce a una particolare regione, quindi si sa già quale sonda usare. Il limite è che, se il fenotipo non si riconduce a sindromi note, non si sa dove cercare: la FISH richiede sempre un indizio.
- Genotype first: si analizza tutto il genoma per identificare il gene causativo e la mutazione che spiega il fenotipo, senza conoscere a priori la regione coinvolta. È l’approccio reso possibile dall’array-CGH e supera i limiti della FISH.
Comparative genomic hybridization (CGH)
L’array-CGH deriva dalla CGH classica, messa a punto per analizzare tessuti tumorali di cui non si avevano metafasi. Si usa la stessa quantità di DNA genomico del tessuto tumorale (verde) e del tessuto di controllo sano (rosso), marcati con fluorocromi di colori diversi, ibridati insieme a normali cromosomi in metafase: si ha un’ibridazione competitiva tra le sequenze marcate in rosso (controllo) e in verde (tumore).
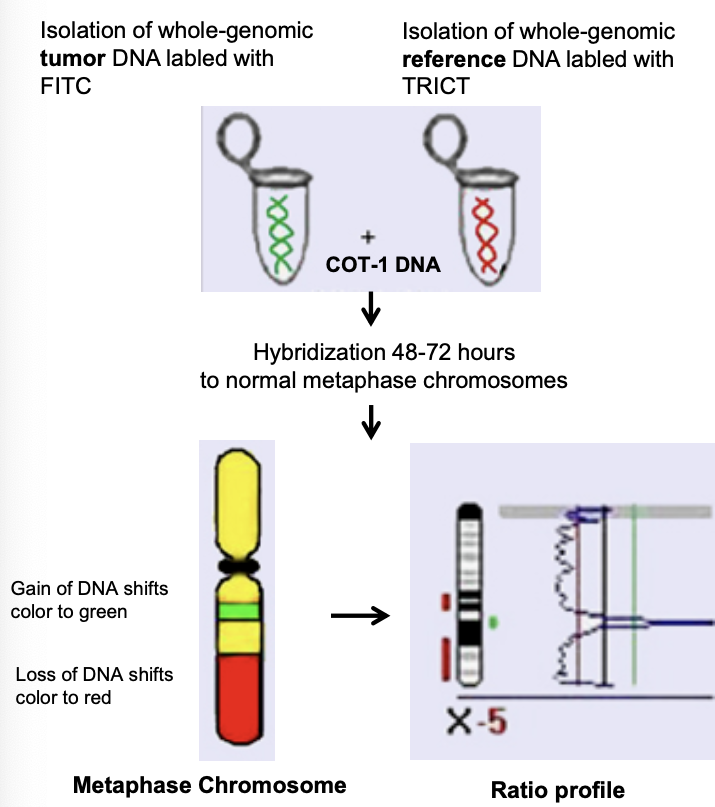
Sulla metafase, se la quantità di DNA dei due tessuti su una sequenza è uguale, l’intensità di ibridazione di verde e rosso è uguale e si ottiene un colore intermedio; se una regione è duplicata o deleta, il segnale di un colore è più alto dell’altro.
Il limite della CGH è la metafase stessa: la risoluzione è di appena 10 Mb (come il bandeggio), quindi sui cromosomi condensati è difficile osservare microdelezioni.
Che cos’è l’array-CGH
Dopo il sequenziamento completo del genoma umano si poterono produrre tutte le sonde specifiche che coprono l’intero genoma e si passò all’uso di un array letto da uno scanner che misura la fluorescenza spot by spot, portando la risoluzione da 10 Mb a poche kb.
In ogni spot dell’array si pone una regione/sequenza del genoma; normalmente si hanno 2 copie per ogni sequenza (perché siamo diploidi). In base alla fluorescenza emessa dalle sonde che si appaiano si stabilisce se ci sono copie in più o in meno della sequenza. A ogni spot corrisponde una posizione genomica: si ottiene quindi il profilo di un intero cromosoma e si individuano delezioni/duplicazioni.
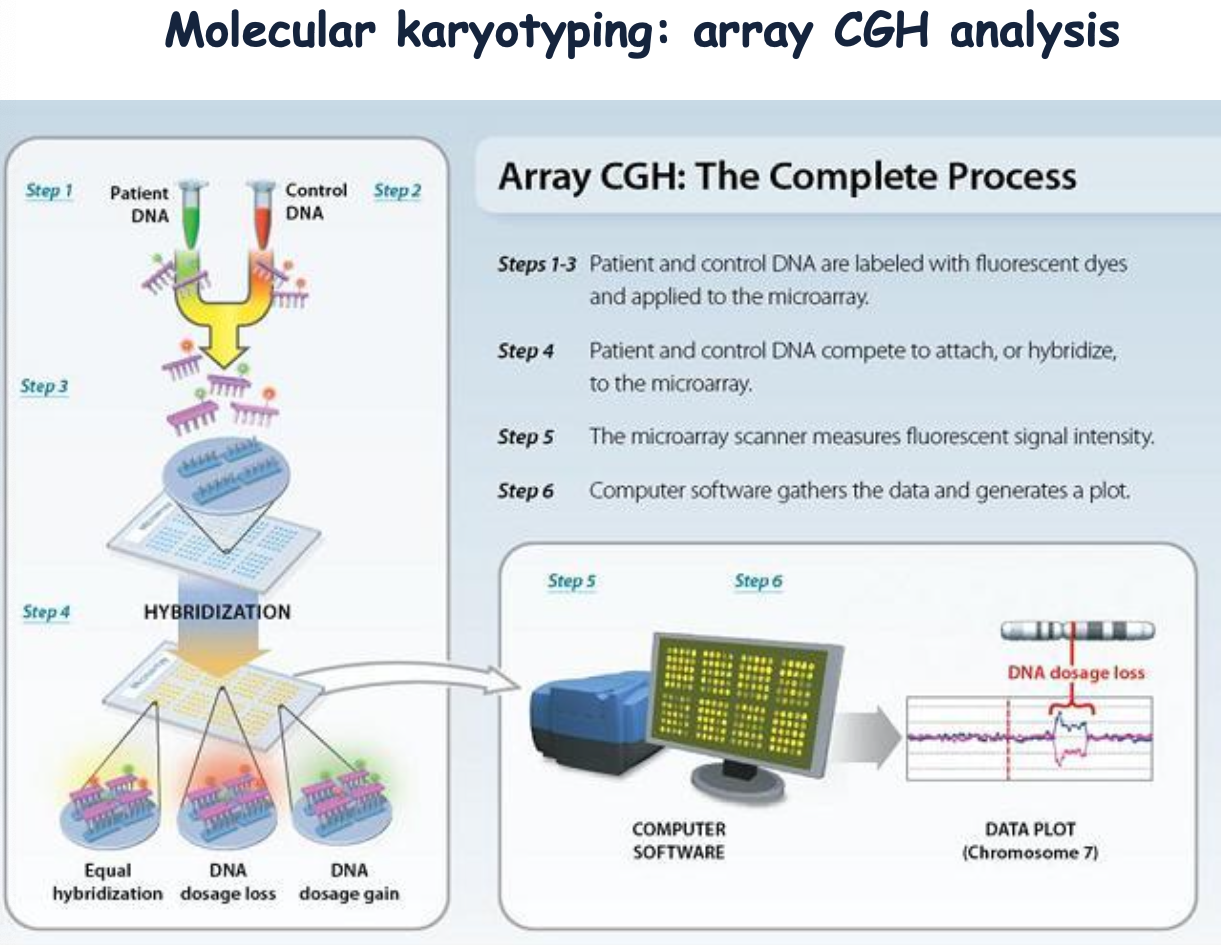
Gli step di preparazione:
- Si marcano le sonde genomiche del paziente e del tessuto di controllo.
- Si ibridano le sonde sui vetrini dei vari spot dell’array.
- Si misura la fluorescenza emessa da ogni spot.
Software specifici collegano l’intensità di fluorescenza alla posizione della sonda, valutando quali geni sono implicati. La risoluzione dipende dal numero di sonde presenti su un vetrino: minore è il numero di sonde ibridate su uno spot, minore è la risoluzione. L’UCSC Genome browser è un sito su cui si trovano tutti i vetrini di array disponibili sul mercato, ciascuno con una certa risoluzione, così da scegliere quello più appropriato alla regione da indagare.
Interpretazione: il log2 ratio
Per fare il profilo dei segnali si calcola il logaritmo in base 2 del rapporto tra l’intensità di fluorescenza del test e quella del reference/controllo:
- Rapporto 1 → log₂ = 0 → regione normale (gli spot sono sullo zero).
- Rapporto ½ → log₂ = −1 → delezione (una sola copia nel campione contro le due del controllo).
- Rapporto 3/2 → log₂ = +0,58 → duplicazione.
- Rapporto che dà log₂ = +1 → quadruplicazione (4 copie nel campione).
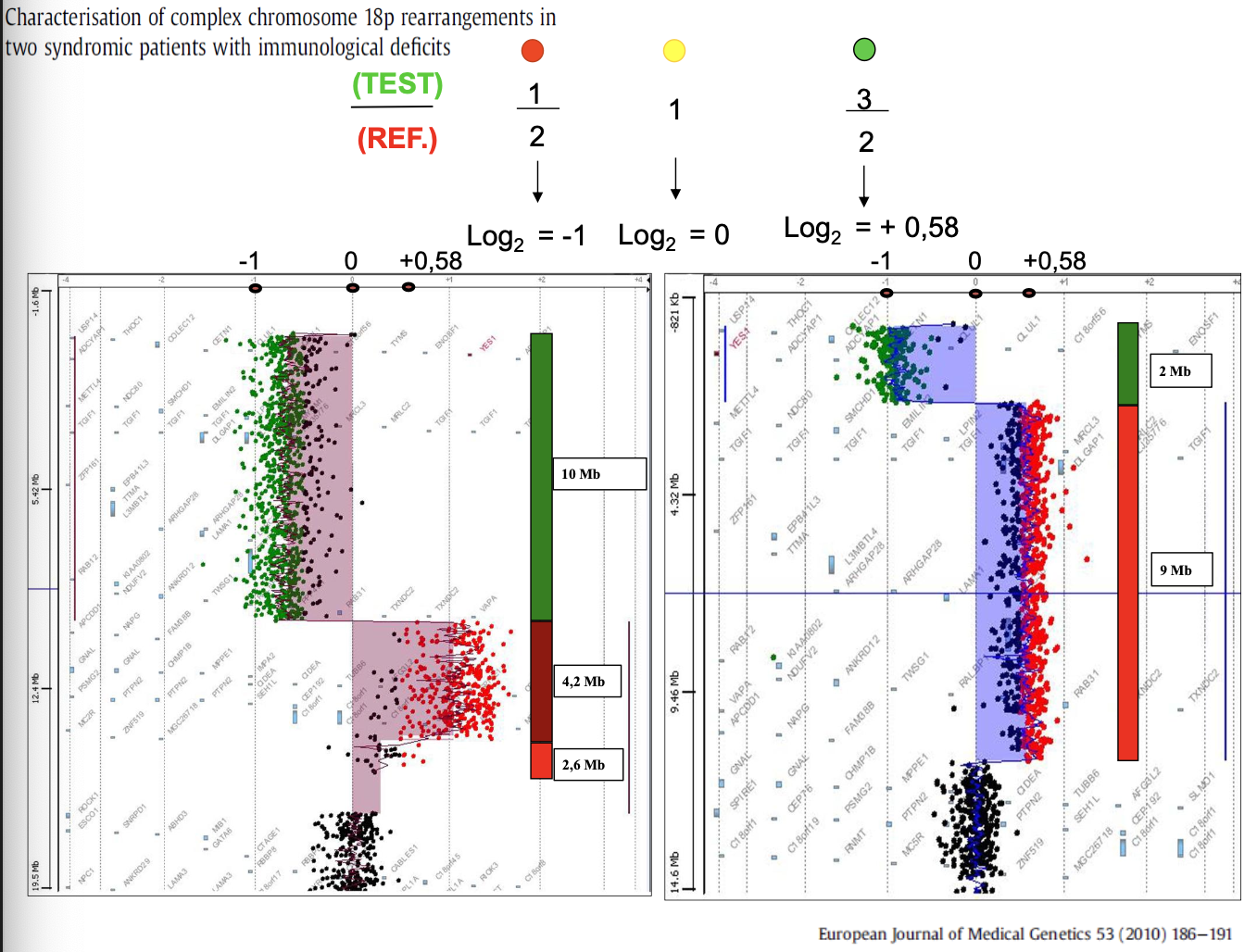
Esempi
- Riarrangiamenti del cromosoma 18. Un soggetto presenta una delezione di una regione di circa 10 Mb, una quadruplicazione di una regione di 4,2 Mb e una duplicazione di una regione di 2,6 Mb.
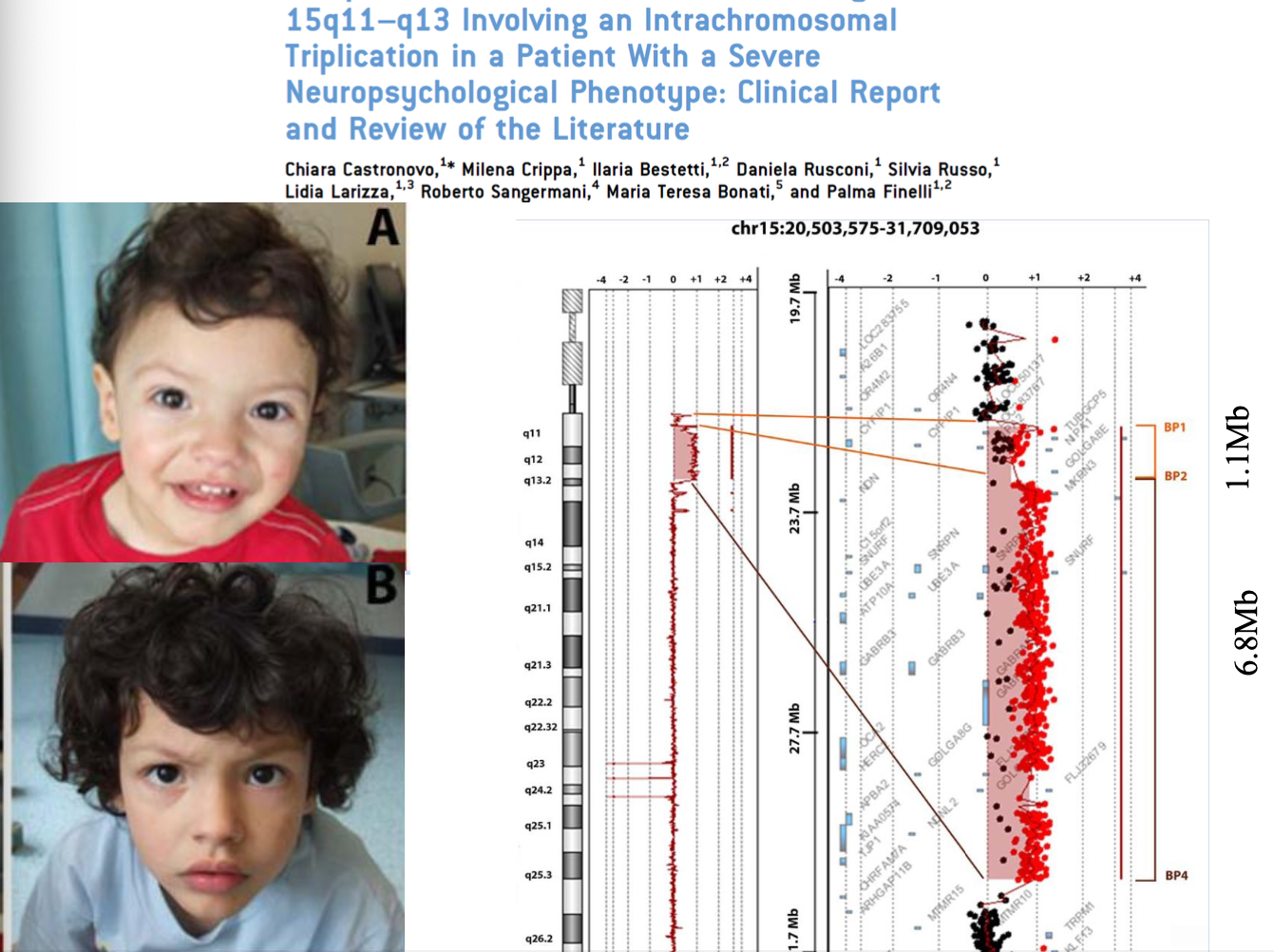
- Cromosoma 15. Un bambino di 9 mesi con fenotipo tipico di un marcatore sovrannumerario del cromosoma 15, che però non presentava tale riarrangiamento. L’array-CGH rivelò sul cromosoma 15 una regione duplicata (1,1 Mb) e una regione triplicata (6,8 Mb).
Limiti
- Non permette di osservare anomalie cromosomiche bilanciate né mosaicismi rappresentati a bassa frequenza.
- Permette di contare il numero di copie di una regione ma non dove questa si trovi.
Esempio (Sotos a mosaico). Un bambino che si credeva avesse la sindrome di Cornelia de Lange non ne presentava il fenotipo (cresceva normalmente, mentre gli affetti hanno ritardo di crescita). Esclusa la Cornelia, l’array-CGH mostrò una piccola delezione in un gene associato alla sindrome di Sotos (che causa overgrowth e non ritardo di crescita), ma il risultato non era chiaro. Ipotizzato un mosaicismo, la FISH dimostrò che il 15% delle cellule del sangue presentava la delezione. La delezione era quindi avvenuta dopo la formazione dello zigote e non era trasmissibile: la madre non rischiava di avere un altro figlio con la stessa condizione.
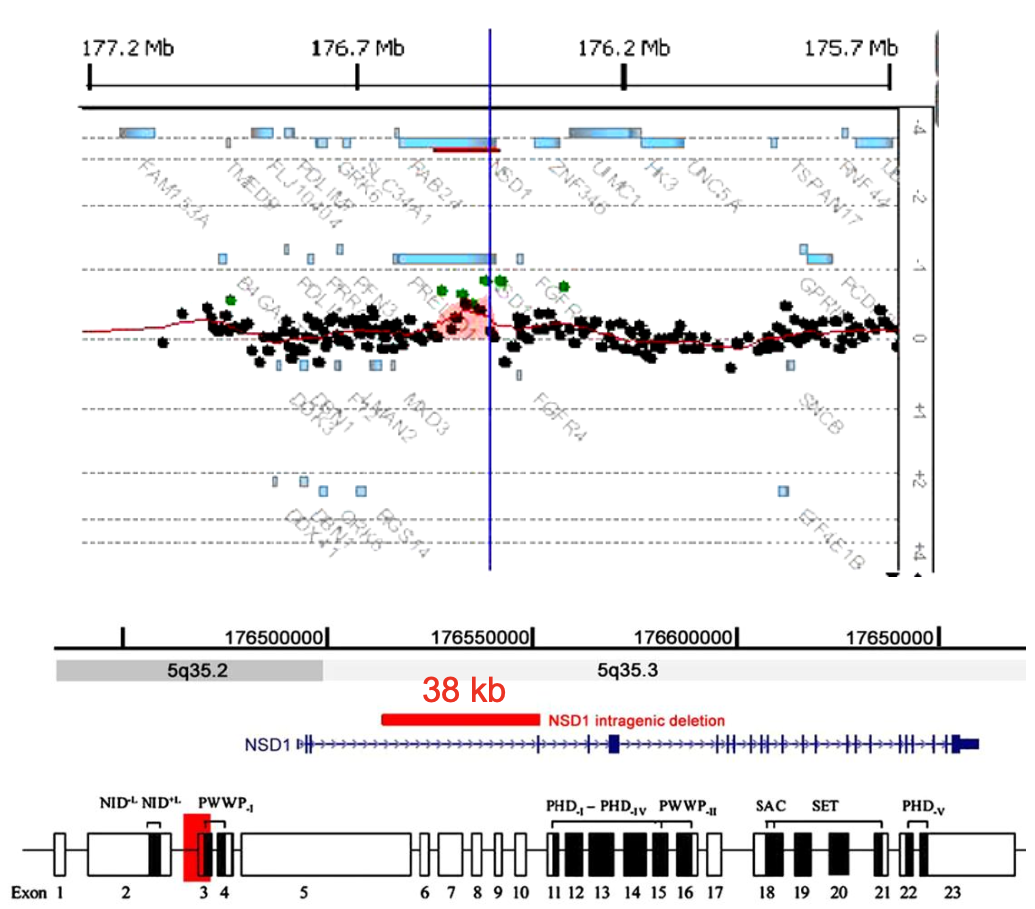
Casi in cui è molto utile
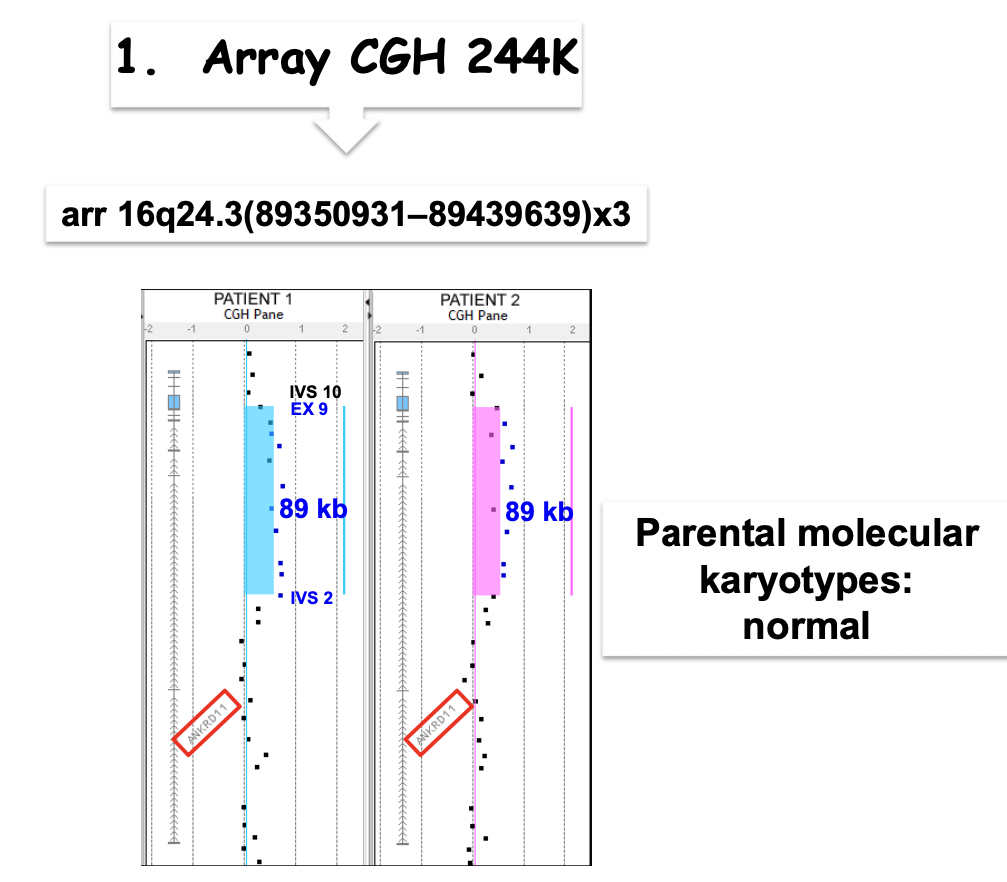
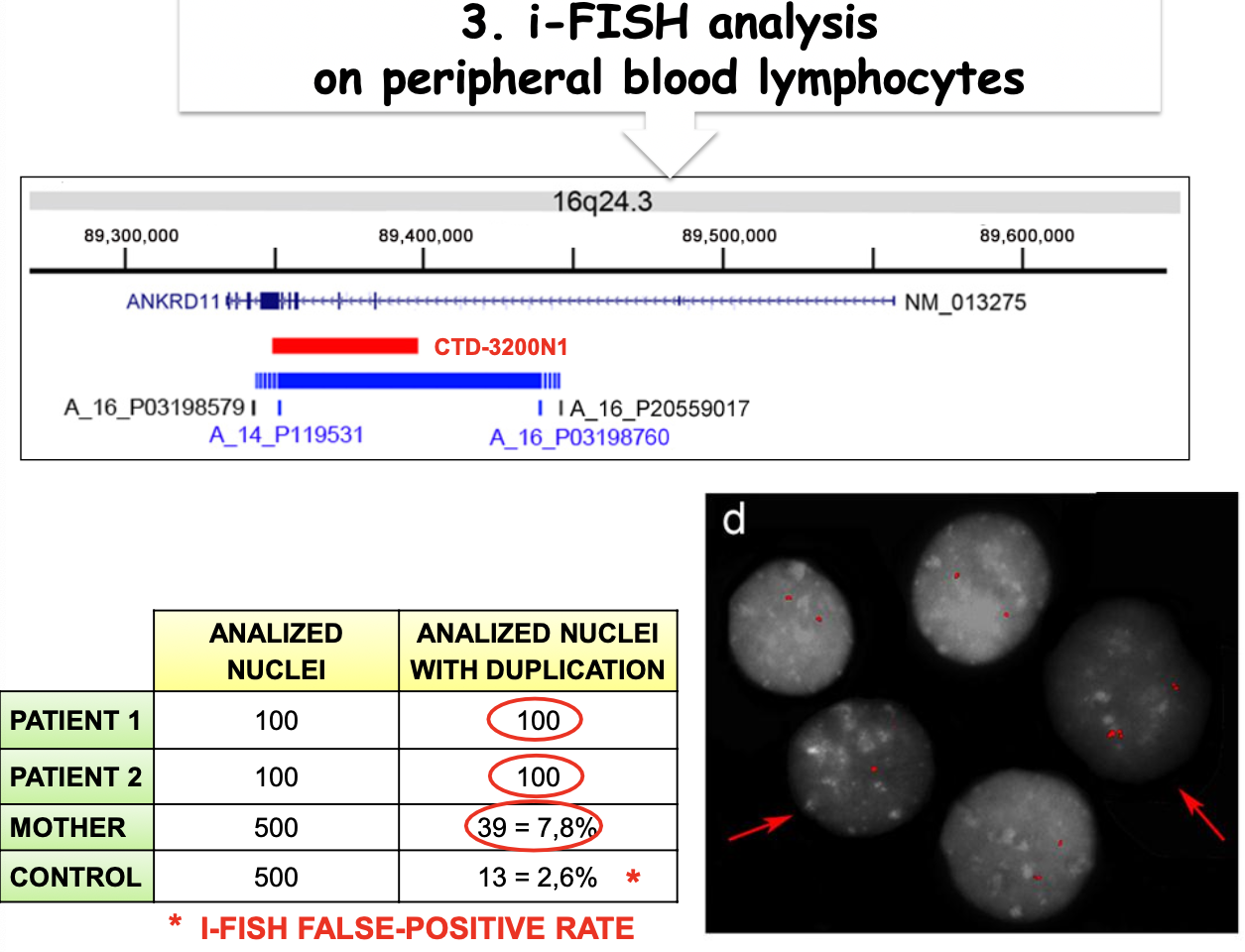
- Mosaicismo germinale e somatico materno (sospetta sindrome KBG). Due fratelli con sospetta sindrome KBG da genitori sani presentavano entrambi una duplicazione della stessa regione del cromosoma 16, evento estremamente improbabile (un singolo riarrangiamento è già molto raro). L’analisi dei microsatelliti nella regione duplicata, eseguita su figli e genitori, mostrò che le copie nei figli derivavano 1 dal padre e 2 dalla madre: era la madre a trasmettere 2 copie. La FISH sul sangue materno rivelò un mosaicismo germinale e somatico, con il 2-6% delle cellule del sangue portatrici della duplicazione.
- Sindrome CHARGE. Sindrome nota da tempo, causa sordità e problemi agli occhi, ma di cui non si conosceva il gene implicato. L’array-CGH su un paziente identificò una delezione in una regione cromosomica; sequenziando i geni della regione deleta si individuò il gene CHD7, confermato anche dall’osservazione di pazienti con mutazione del gene ma senza la regione deleta. L’array-CGH permette quindi di identificare il gene causativo di una sindrome nota da tempo.
🔗 Collegamenti
- Citogenetica Molecolare — 📋 fa parte di
- FISH — 🔄 confronto
- Patologie Genomiche — 🔬 reperto diagnostico
- Copy Number Variation — ⬇️ conseguenza
- Identificazione di Geni delle Malattie — 🔗 stesso meccanismo / stessa via
- Mosaicismo — 🔬 reperto diagnostico