Sindrome di Smith-Magenis
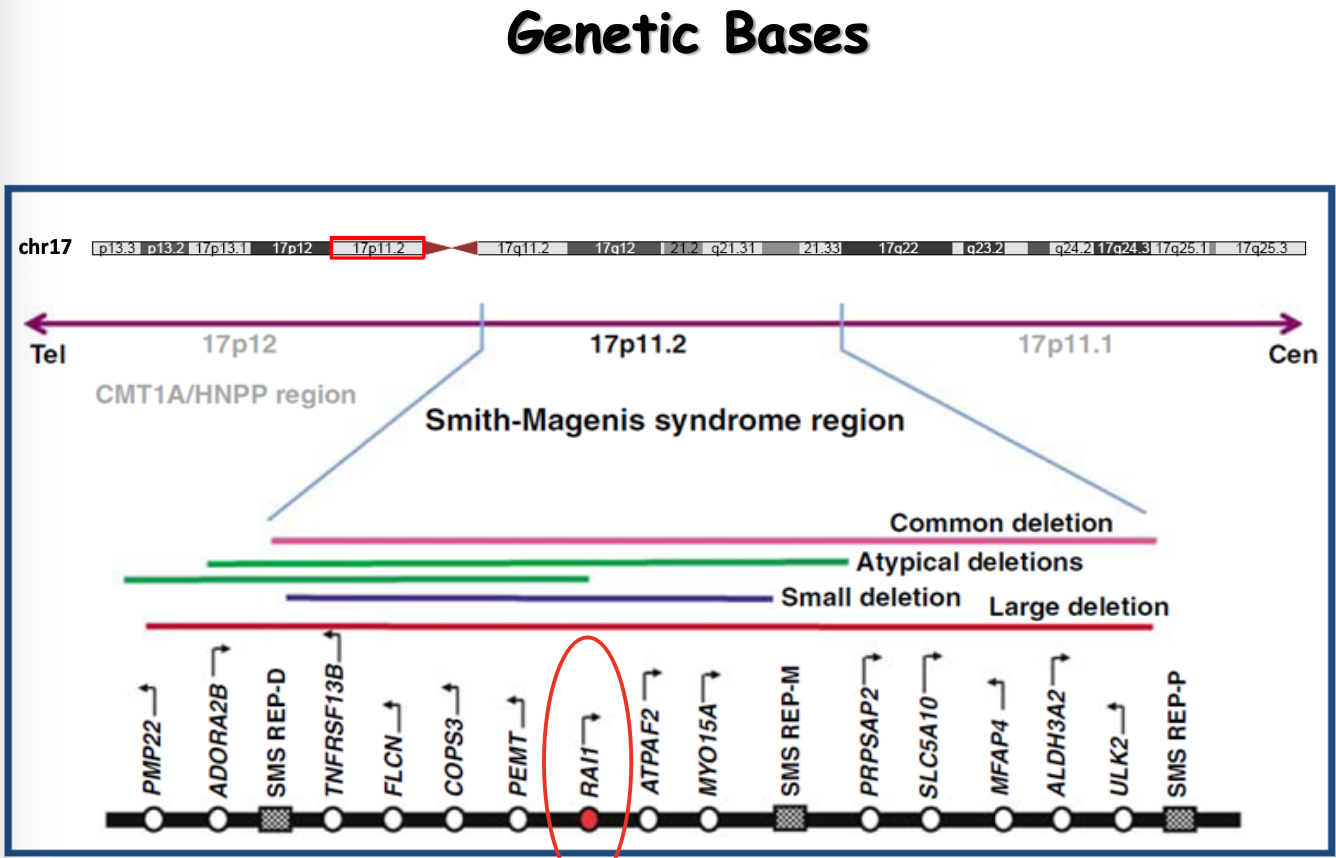
La sindrome di Smith-Magenis (SMS) è una patologia genomica causata dalla delezione di una regione del cromosoma 17, limitata alle estremità da duplicazioni segmentali.
Relazione con Charcot-Marie-Tooth
La regione che, deleta, causa la SMS si trova accanto al gene PMP22. La duplicazione del gene PMP22 (coinvolto nella formazione della mielina) causa invece la malattia di Charcot-Marie-Tooth (CMT), una malattia neuromuscolare periferica. Un riarrangiamento di questa regione cromosomica può quindi dare due patologie diverse a seconda che si abbia delezione (SMS) o duplicazione (CMT).
Quadro clinico
- Dismorfismi scheletrici e facciali peculiari.
- Obesità e bassa statura.
- Caratteristiche psichiche particolari: individui iperattivi, abbastanza aggressivi, con tendenze autolesionistiche.
- Disturbi del sonno.
Gene RAI1
La regione deleta contiene diversi geni, ma il gene RAI1 è l’unico gene causativo: è solo per la sua perdita che si manifesta la sindrome. La certezza deriva dal fatto che, in tutti i pazienti osservati, pur variando la lunghezza della delezione, RAI1 viene sempre deleto o subisce comunque una mutazione puntiforme. La sindrome è dovuta:
- per il 90% a delezione della regione del cromosoma 17 che include RAI1;
- per il 10% a mutazione puntiforme o microdelezione all’interno del gene RAI1.
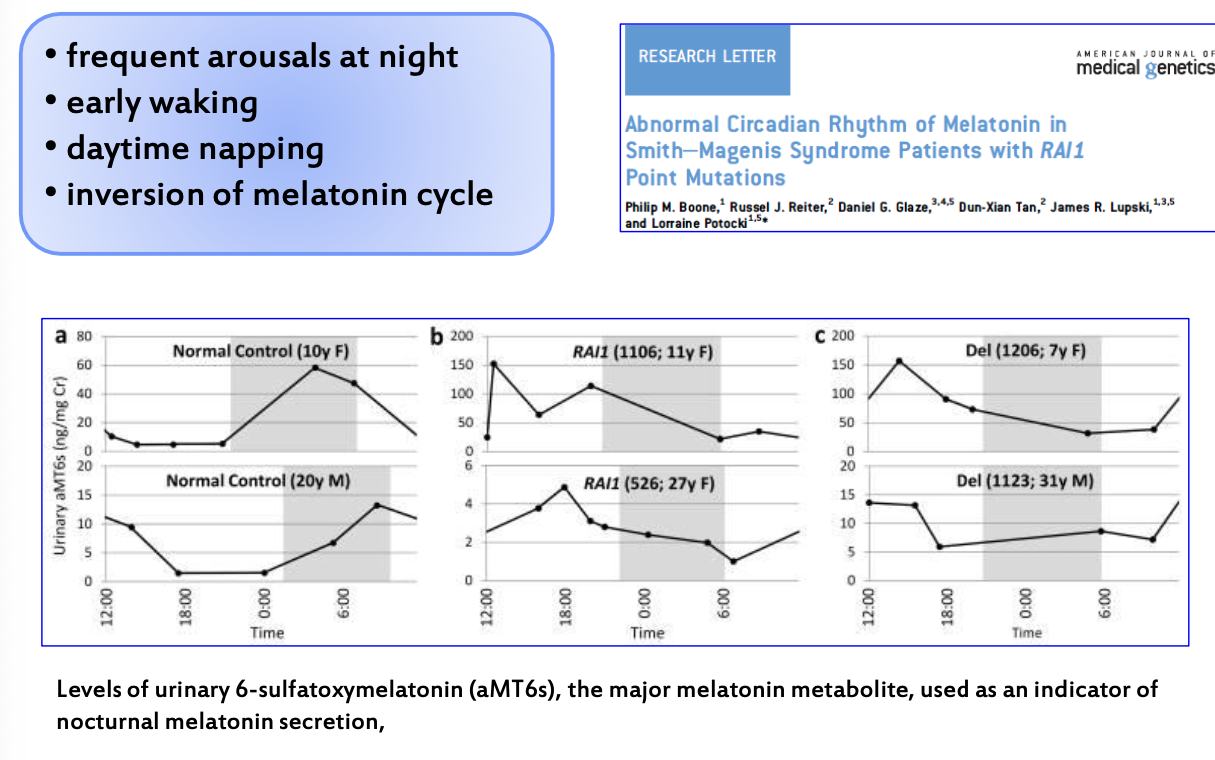
Disturbi del sonno e ritmo circadiano
I disturbi del sonno sono dovuti a un’inversione del ritmo circadiano. Ogni cellula riconosce la fase del giorno producendo specifiche proteine nelle diverse fasi; questa regolazione è mediata dalla melatonina. In un individuo normale la melatonina è prodotta e rilasciata tra mezzanotte e le 6:00 del mattino. Nei pazienti con SMS l’aumento di produzione e rilascio di melatonina avviene intorno a mezzogiorno, mentre la concentrazione diminuisce durante la notte.
Sindrome di Potocki-Lupski
Se la regione normalmente deleta nella SMS va incontro a duplicazione, si manifesta un’altra patologia, la sindrome di Potocki-Lupski (PTLS).
Diagnosi della SMS
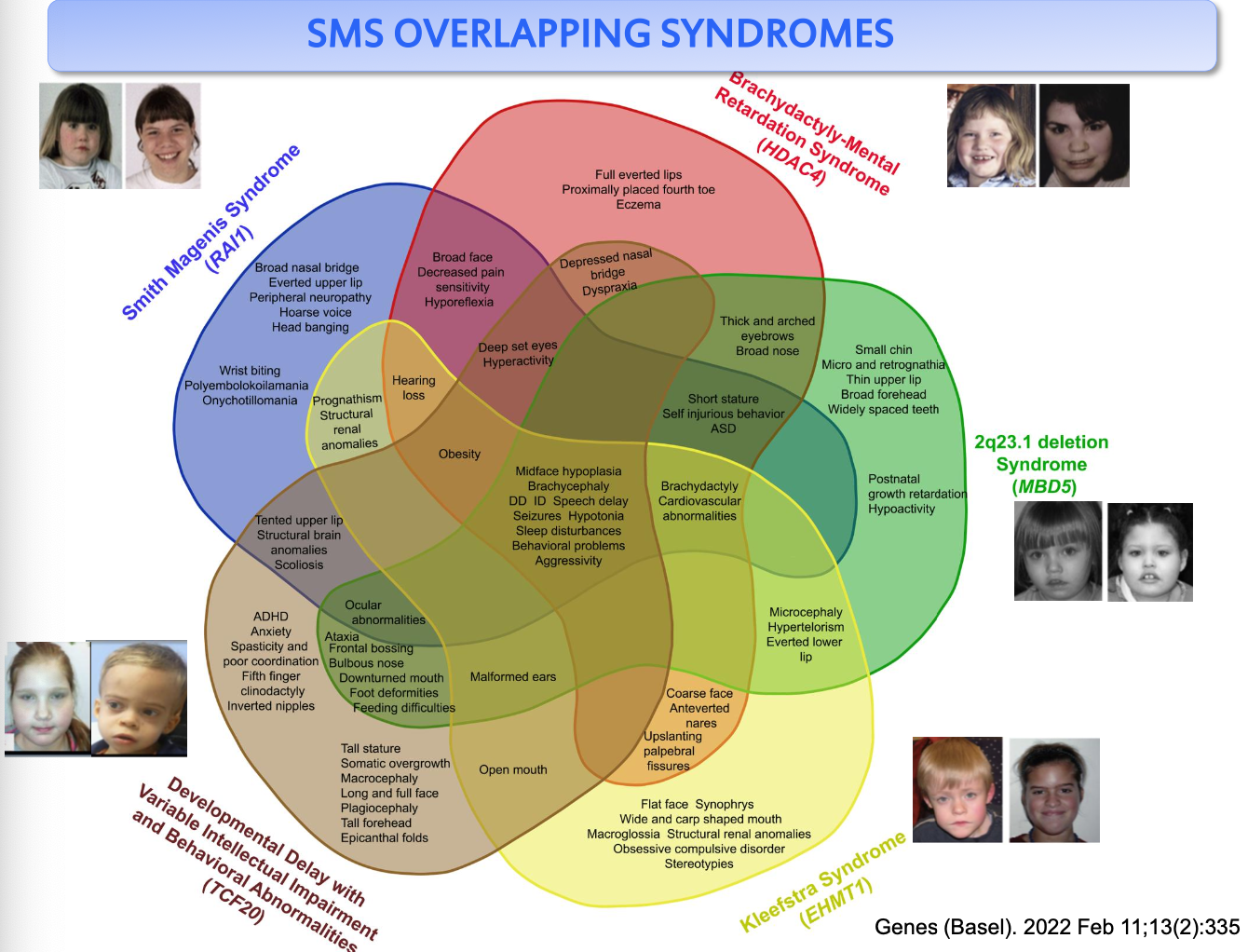
Di fronte a un fenotipo compatibile con la SMS, solo nel 50% dei casi si riesce a confermare la diagnosi; nell’altro 50% non si trovano né mutazioni in RAI1 né microdelezioni. Questa omologia di fenotipo tra individuo affetto e non affetto è dovuta all’esistenza di geni che regolano RAI1: se uno di questi geni regolatori non è espresso, si ha una down-regolazione di RAI1 e un fenotipo analogo a quello da RAI1 non funzionale. Tutti questi geni correlati a RAI1 (tra cui HDAC4) entrano in diagnosi differenziale con RAI1.
🔗 Collegamenti
- Patologie Genomiche — 📋 fa parte di
- Duplicazioni Segmentali — ⬆️ causa
- Delezione Cromosomica — ⬆️ causa