Patogenesi dell’Autoimmunità
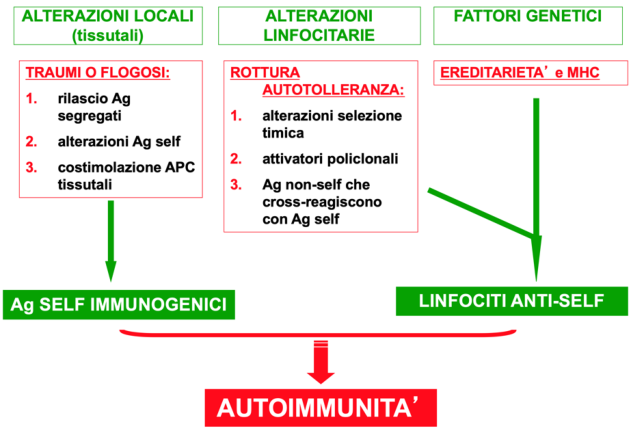
La patogenesi dell’autoimmunità è complessa e coinvolge fattori che alterano l’espressione delle proteine nei tessuti, la configurazione degli antigeni, particolari molecole MHC e alterazioni della selezione timica. Schematicamente, intervengono alterazioni locali tissutali, alterazioni linfocitarie e fattori genetici: quando le alterazioni linfocitarie si associano ai fattori genetici si ha l’attivazione dei linfociti anti-self e l’insorgenza dell’autoimmunità.
Alterazioni locali tissutali
Rendono il self immunogenico, cioè riconoscibile:
- Rilascio di Ag segregati: rottura di una barriera anatomica.
- Alterazioni degli Ag self.
- Costimolazione su cellule tissutali: in seguito a infiammazione, le cellule tissutali possono esprimere molecole di costimolo e permettere l’attivazione dei linfociti.
Alterazioni linfocitarie
- Alterazioni della selezione timica.
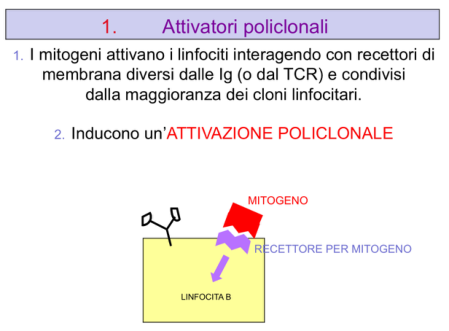
- Attivatori policlonali: attivano molteplici cloni di linfociti T e B, tra cui possono trovarsi cloni anti-self.
- Ag non-self che cross-reagiscono con Ag self.
Fattori genetici
Coinvolgono l’ereditarietà e le MHC. Alcuni aplotipi HLA predispongono a determinate malattie autoimmuni.
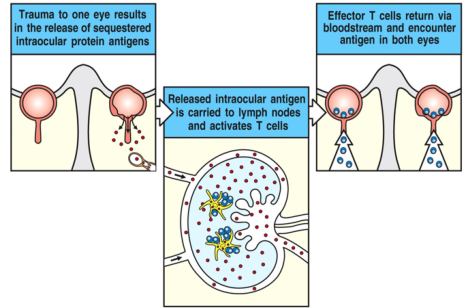
Ag segregati
Se si rompe la barriera anatomica, i linfociti incontrano antigeni self mai visti prima e si attiva una risposta infiammatoria. La classica pallonata nell’occhio rompe la barriera dietro la cornea, liberando antigeni oculari che vengono aggrediti dai linfociti: ne deriva un’oftalmite. Anche rompendo la barriera in un solo occhio si colpisce pure l’altro: si parla di oftalmite simpatica perché interessa entrambi gli occhi.
Ag ignorati
Sono antigeni a cui i linfociti hanno accesso, ma che sono espressi in bassissima quantità: i linfociti li vedono ma non li attaccano. In alcuni casi, per motivi non noti, la loro quantità aumenta e scatta l’aggressione da parte del sistema immune.
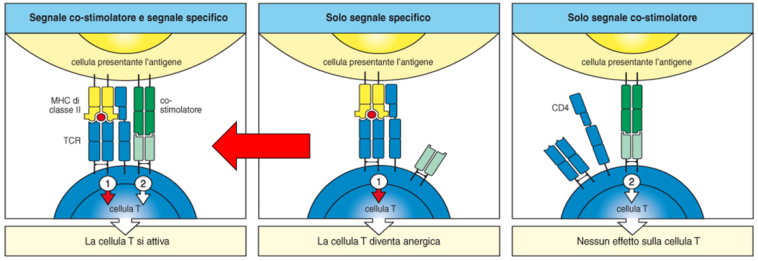
Costimolazione
Nelle cellule dei tessuti periferici non sono presenti molecole di costimolo: nel fegato, ad esempio, l’antigene self è presentato ma, in assenza di costimolo, il linfocita non si attiva pur riconoscendolo (anergia). In seguito a infiammazione o infezioni, le citochine prodotte inducono le cellule tissutali ad aumentare l’espressione di HLA e di molecole di costimolo (CD40, CD80, CD86). Le APC tissutali diventano così in grado di attivare i linfociti T contro il self.
Modelli sperimentali
- Polli di ceppo obeso (OS): producono auto-anticorpi anti-tiroide che ne provocano la distruzione. Se si rimuove la borsa di Fabrizio (sito di maturazione dei linfociti B negli uccelli, equivalente del midollo osseo umano), la tiroidite è meno grave, perché ci sono meno auto-Ab anti-tiroide. Se si rimuove il timo, la malattia peggiora, perché agli auto-anticorpi si aggiunge la riduzione dei Treg. (Dal nome della borsa di Fabrizio deriva il nome “linfociti B”.)
- Attivazione policlonale nel topo: iniettando attivatori policlonali come gli LPS (lipopolisaccaride) si attivano moltissimi cloni di linfociti T e B, compresi cloni diretti contro il self, con sviluppo di patologia autoimmune. Nell’uomo non esiste una patologia autoimmune chiaramente correlata all’alterazione dei Treg o del bilancio Th1/Th2; in teoria un superantigene potrebbe avere effetto analogo, ma non ci sono casi documentati.
🔗 Collegamenti
- Malattie Autoimmuni — 📋 quadro generale dell’autoimmunità
- Mimetismo Molecolare — 🔗 cross-reattività Ag non-self/self come meccanismo patogenetico
- Associazione HLA-Malattie — 🔗 fattori genetici (HLA) nella predisposizione
- Mitogeni e Attivatori Policlonali — 🔗 attivazione policlonale di cloni anti-self
- Segnali Costimolatori e Sinapsi Immunitaria — 🔗 ruolo del costimolo nell’attivazione T
- Fattori Predisponenti dell’Autoimmunità — ⬆️ condizioni che favoriscono questi meccanismi