Aterosclerosi
Definizione
L’ateroma è una lesione cronica infiammatoria della parete delle arterie di medio e grosso calibro. È paragonabile a un granuloma cronico, ma al posto di un agente infettivo vi è l’accumulo di lipidi, in particolare LDL ossidate. Si forma nella tonaca intima, sotto l’endotelio, e non sporge direttamente nel lume. Condizione necessaria è l’iperlipidemia, soprattutto LDL elevate.
L’aterosclerosi, attraverso gli eventi cardiovascolari, è la principale causa di morte; rientra nel quadro della Sindrome Metabolica.
Ateroma vs trombo
- Ateroma: cresce lentamente sotto l’endotelio, restringendo progressivamente il lume (stenosi). Di per sé non si stacca.
- Trombo: coagulo acuto formato dentro il lume (piastrine, GR, GB), che può staccarsi generando un embolo.
L’ateroma può però, rompendo l’endotelio che lo ricopre, innescare un trombo sovrastante.
Eziologia — fattori di rischio
Il fattore fondamentale è l’ipercolesterolemia (LDL alte): l’infiammazione da sola non basta, serve sempre ipercolesterolemia. Altri fattori, spesso associati alla sindrome metabolica:
- Ipertensione → danno meccanico all’endotelio.
- Iperglicemia / diabete → glicazione, ROS, stress ossidativo.
- Fattori emodinamici → flusso turbolento, biforcazioni.
- Tossine, virus, fumo → danno diretto alle cellule endoteliali.
- Omocisteinemia → aumenta lo stress ossidativo e riduce il glutatione.
Ruolo dell’endotelio
In condizioni normali l’endotelio mantiene fluido il sangue (fattori anticoagulanti) e regola la vasomotilità. Se danneggiato: aumenta la permeabilità, richiama monociti e piastrine, libera citochine proinfiammatorie e perde le funzioni anticoagulanti.
Alterazioni emodinamiche
Il flusso turbolento (biforcazioni arteriose, ipertensione) danneggia più facilmente l’endotelio. Zone critiche sono carotidi e coronarie: se ostruite, rischio di ictus o infarto.
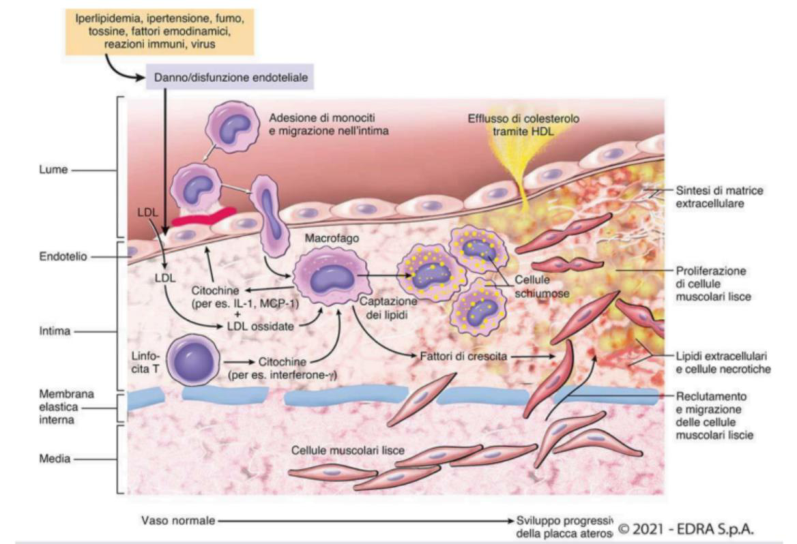
Patogenesi — formazione della placca
- Danno/disfunzione endoteliale (ROS, ipertensione, diabete, fumo) in presenza di LDL alte.
- Le LDL passano nell’intima, sotto l’endotelio, dove vengono ossidate dai ROS prodotti dalle cellule infiammatorie e dall’endotelio attivato.
- Reclutamento di monociti, che aderiscono, fanno diapedesi e diventano macrofagi.
- I macrofagi riconoscono e fagocitano le LDL ossidate tramite recettori scavenger, trasformandosi in cellule schiumose. Si formano le strie lipidiche (chiazze giallastre sottoendoteliali che precedono l’ateroma). Alcuni macrofagi muoiono e rilasciano lipidi, amplificando la lesione.
- I macrofagi rilasciano citochine che richiamano linfociti T e cellule muscolari lisce.
- Le cellule muscolari lisce della tonaca media migrano nell’intima, si differenziano in miofibroblasti e depositano collagene.
- Si forma la placca fibro-adiposa (lipidi + cellule schiumose + collagene), con calcificazioni legate al deposito di collagene.
- Crescita lenta → stenosi cronica.
- Rottura dell’endotelio → trombosi acuta → infarto/ictus.
Il coinvolgimento delle cellule muscolari lisce spiega perché la malattia colpisca solo le arterie di medio e grosso calibro, dotate di una tonaca media sviluppata.
Ingresso delle LDL
- Ipotesi classica: trasporto tramite caveole (endocitosi aspecifica).
- Ipotesi più recente: trasporto mediato dal recettore SR-B1 (specifico).
Bloccare questo ingresso potrebbe impedire la formazione della placca.
Polarizzazione dei macrofagi nella placca
I linfociti T modulano i macrofagi (vedi Polarizzazione dei Macrofagi (M1-M2)):
- M1 (attivati da IFN-γ dei Th1): pro-infiammatori, fagocitosi → placca più fragile.
- M2 (attivati da IL-4 e IL-10 dei Th2): effetto riparativo, stimolano fibroblasti e neoangiogenesi, rilasciano fattori di crescita che richiamano le cellule muscolari lisce → placca più stabile.
Ruolo dei linfociti e delle metalloproteasi
I linfociti T stimolano i macrofagi a produrre metalloproteasi che degradano il collagene, rendendo la placca più fragile e a rischio di rottura. Se prevalgono i segnali fibrogenici (IL-4, IL-10) si deposita più collagene → placca più stabile.
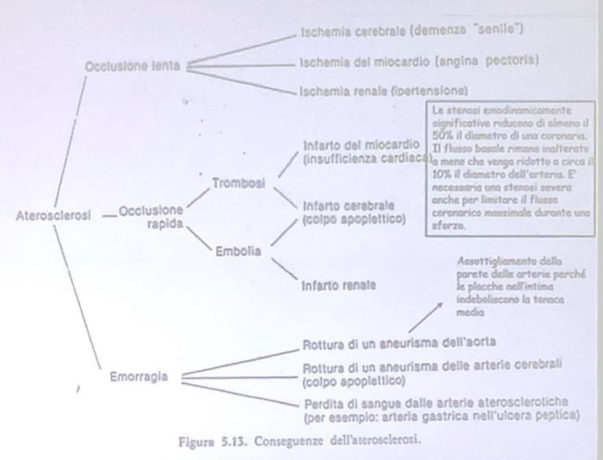
Quadro clinico — complicanze
La placca, costituita da un cappuccio fibroso e un centro sclerotico, cresce lentamente causando stenosi progressiva e ischemia cronica (angina, demenza vascolare, ipertensione renale).
Se l’endotelio che la ricopre si rompe, si forma un trombo sovrastante (piastrine + fibrina + GB + GR) che può occludere completamente il vaso → infarto (coronarie) o ictus (carotidi). Da un trombo può staccarsi un embolo che occlude vasi più piccoli a valle:
- nelle vene → embolia polmonare; nelle arterie → ischemia degli organi irrorati.
- nell’aorta la placca può causare dilatazione/aneurisma, con rischio di rottura ed emorragia intraplacca (rottura dei neovasi da neoangiogenesi).
Diagnosi
Screening con ecocolordoppler dei tronchi sovraortici / carotideo: le carotidi fungono da “specchio” della situazione vascolare generale e le placche vi sono accessibili.
Terapia
- Statine: bloccano la sintesi epatica di colesterolo → riducono le LDL → rallentano la crescita della placca.
- Angioplastica con palloncino o altre procedure interventistiche se la placca occupa più del 60-70% del lume.
(Integrazione da: sbobina 270-271)
🔬 Reperto istologico — stadi della placca
Lesione iniziale (stria lipidica). Intima ispessita, media ancora normale; aree chiare dove erano presenti lipidi (dissolti in preparazione) e cellule schiumose (foam cells) = macrofagi o cellule muscolari lisce che hanno fagocitato lipidi (citoplasma vacuolizzato e chiaro). Prevalgono lipidi, macrofagi, cellule muscolari lisce e linfociti T → lesione precoce e reversibile, placca ancora “molle”.
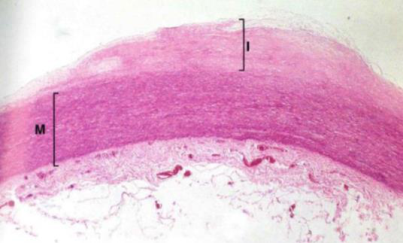
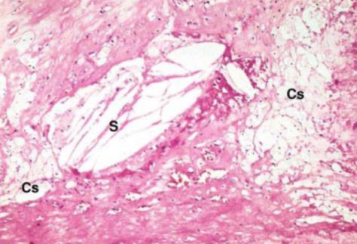
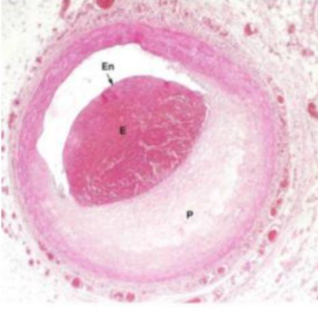
Placca fibro-lipidica. Zona lipidica centrale con un cappuccio fibroso (fibroblasti + collagene) sotto l’endotelio. La tonaca media inizia a ridursi di spessore → perdita di elasticità → fase stabile, con rischio di dilatazioni/aneurismi.
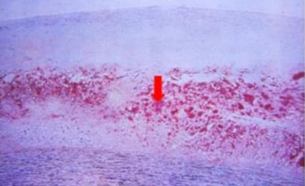
Progressione. Cappuccio fibroso più spesso, componente lipidica più estesa, tonaca muscolare ulteriormente assottigliata; la placca sporge sempre più nel lume, riducendolo.
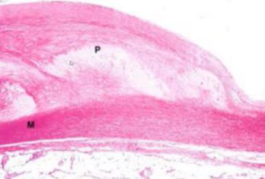
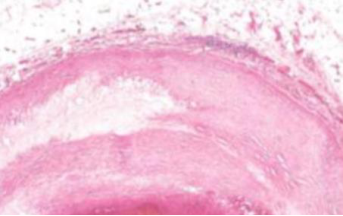
Rottura e trombosi. Fessurazioni e rotture superficiali; nelle zone rotte si forma un trombo (massa scura) che può occludere il vaso → ischemia o infarto.
Placca stabile vs instabile. La stabile ha cappuccio fibroso spesso e pochi lipidi (restringe il lume ma non si rompe facilmente); l’instabile ha cappuccio sottile, molti lipidi e cellule schiumose, si rompe facilmente esponendo materiale trombogenico → trombi.
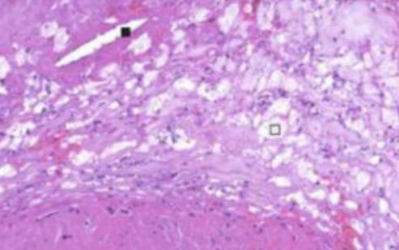
Ostruzione del vaso. La placca eccentrica occupa gran parte del lume. Colorazione di Van Gieson: rosso = collagene (fibrosi), giallo = muscolatura liscia.
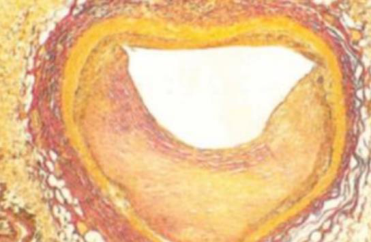
Placca con emorragia. Eritrociti (zone rosse) all’interno della placca → rottura interna e sanguinamento intraplacca, tipico delle placche instabili.
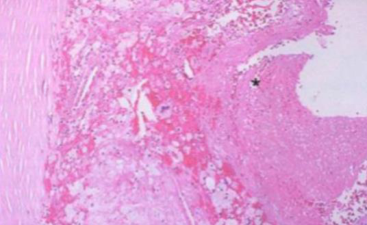
Ateroma complicato. Grande accumulo lipidico, trombo sovrapposto ed emorragia intraplacca; lume quasi completamente occluso → ischemia o infarto nei tessuti irrorati.
🔗 Collegamenti
- Sindrome Metabolica — ⬆️ contesto eziologico principale
- Metabolismo delle Lipoproteine — 🔗 origine delle LDL aterogene
- Trombosi — ⬇️ complicanza acuta su placca rotta
- Infarto del Miocardio — ⬇️ conseguenza su coronarie
- Polarizzazione dei Macrofagi (M1-M2) — 🔗 determina stabilità della placca
- Infiammazione Cronica — 🔗 natura della lesione
- Stress Ossidativo — ⬆️ ossidazione delle LDL