Amiloidosi
Definizione
L’amiloidosi (o beta-fibrillosi) è una malattia da accumulo caratterizzata da depositi extracellulari (interstiziali) di proteine fibrillari anomale, dette amiloidi, che danneggiano tessuti e organi. I depositi sono insolubili, rigidi e alterano la funzione dell’organo colpito.
Non è una singola malattia, ma un insieme di sindromi diverse con cause differenti.
Eziologia
- Genetiche → mutazioni che portano a proteine anomale (es. transtiretina mutata).
- Infiammazione cronica → iperproduzione di proteine infiammatorie che non vengono degradate.
- Tumori (es. mieloma multiplo) → produzione anomala di immunoglobuline o loro frammenti.
Struttura dell’amiloide
Le proteine amiloidi assumono una conformazione anomala ricca di foglietti β: questa struttura favorisce l’aggregazione in fibrille insolubili. Anche proteine normali possono cambiare conformazione e formare foglietti β, diventando insolubili.
I depositi amiloidi contengono:
- 90% proteine fibrillari (la componente patologica vera e propria).
- 10% glicoproteine (che stabilizzano il deposito).

Patogenesi
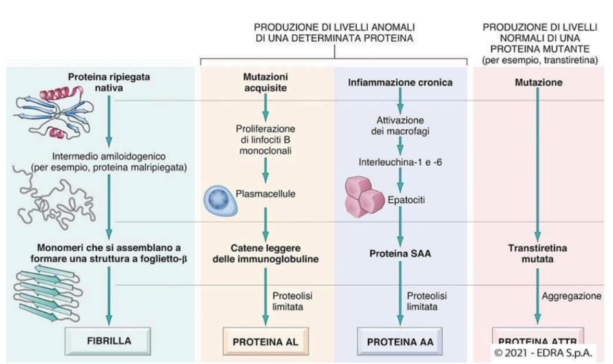
Quattro meccanismi di formazione dell’amiloide:
- Da infiammazione cronica: i macrofagi producono IL-1 e IL-6 → stimolano il fegato a produrre SAA. Se la degradazione di SAA è insufficiente → si accumula e precipita come amiloide AA. → Amiloidosi AA (Secondaria)
- Da mutazioni acquisite (tumorali): le plasmacellule tumorali (mieloma) producono catene leggere di Ig in eccesso → amiloide AL. Le proteine sono normali, ma in quantità anormali. → Amiloidosi AL (Primaria)
- Da mutazioni genetiche: si producono proteine mutate e instabili (es. transtiretina mutata) che si depositano anche se in quantità normali → amiloidosi ereditaria.
- Da proteine native mal ripiegate: anche proteine normali possono cambiare conformazione e formare foglietti β, diventando insolubili e depositandosi.
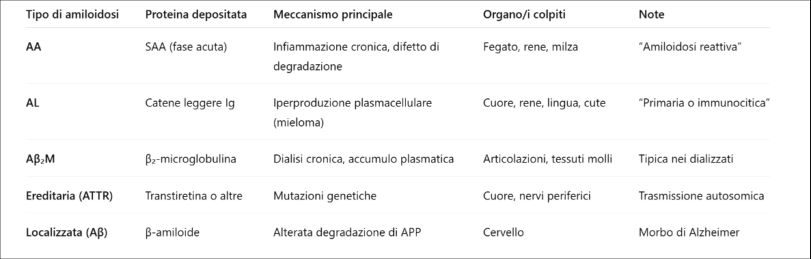
Tipi principali (secondo la proteina coinvolta)
- Amiloide AL (catene leggere): deriva da catene leggere delle immunoglobuline prodotte in eccesso da un clone di plasmacellule. Amiloidosi primaria (immunocitica). → Amiloidosi AL (Primaria)
- Amiloide AA: deriva dal precursore SAA (Serum Amyloid A), proteina di fase acuta prodotta dal fegato durante l’infiammazione. Amiloidosi secondaria/reattiva (da infiammazione cronica). → Amiloidosi AA (Secondaria)
- Amiloide Aβ (β-amiloide): deriva dalla proteina APP (Amyloid Precursor Protein), si deposita nel cervello → tipico del morbo di Alzheimer. Amiloidosi localizzata (cerebrale). → Amiloidosi Cerebrale (Alzheimer)
Classificazione
In base alla localizzazione:
- Sistemica → coinvolge più organi contemporaneamente (rene, cuore, fegato…).
- Localizzata → deposito in un solo organo (es. Alzheimer nel cervello).
In base alla causa (eziologia):
- Ereditaria (genetica) → proteine mutate (es. transtiretina).
- Acquisita:
- Reattiva (secondaria) → infiammazione cronica.
- Primaria (immunocitica) → mieloma o plasmocitosi.
Organi più colpiti
- Rene → sindrome nefrosica e insufficienza renale.
- Cuore (miocardio) → cardiomiopatia restrittiva.
- Fegato → epatomegalia.
- Milza → splenomegalia.
Amiloidosi localizzate
Limitate a un singolo organo, spesso legate a tumori endocrini o neuroendocrini:
- Carcinoma midollare della tiroide → iperproduzione di precursore della calcitonina → accumulo amiloide locale.
- Insulinoma (tumore pancreatico) → iperproduzione di amilina (co-secretata con insulina) → deposito amiloide pancreatico.
- Prioni → encefalopatia.
La forma localizzata cerebrale più importante è quella dell’Alzheimer. → Amiloidosi Cerebrale (Alzheimer)
Diagnosi
La diagnosi è spesso difficile perché l’amiloidosi può colpire diversi organi. Si sospetta quando:
- la clinica è atipica o non spiegabile con altre patologie;
- si osservano disfunzioni d’organo multiple (rene, cuore, fegato…).
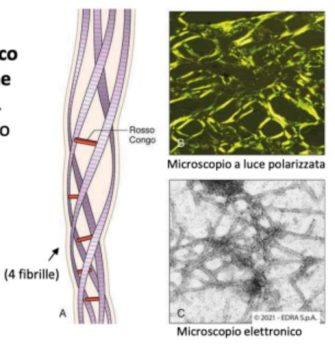
Conferma diagnostica: dimostrazione del deposito amiloide nel tessuto con colorazioni specifiche, in particolare il Rosso Congo, che mostra birifrangenza verde in luce polarizzata.
Terapia
Terapie emergenti anti-amiloide, ancora in fase di sviluppo:
- Stimolare i macrofagi a rimuovere i depositi amiloidi.
- Utilizzare anticorpi specifici diretti contro le proteine amiloidi.
(Integrazione da: sbobina 267-269)
🔬 Riconoscimento istologico dell’amiloide
Colorazioni:
- Ematossilina-eosina (H&E) → l’amiloide appare rosa pallido, amorfa ed extracellulare, ma non distinguibile da collagene o altre proteine.
- Rosso Congo → colorazione specifica: amiloide rosso-arancio in luce normale; in luce polarizzata mostra la caratteristica rifrangenza verde-mela (dato diagnostico).
- Rosso Sirio, Lugol (iodio iodurato), acido solforico, Tioflavina-T → evidenziano l’amiloide in modo specifico o fluorescente (verde sotto UV).
Effetti tissutali generali. L’amiloide si deposita nell’interstizio e lungo le membrane basali, causando ispessimento, compressione dei vasi e ridotta perfusione → ischemia. Le cellule circostanti vanno incontro a sofferenza, atrofia e danno funzionale. Macroscopicamente l’organo può essere aumentato di volume e di consistenza gommosa (fasi iniziali) o atrofico (fasi tardive ischemiche).
Diagnosi pratica. Oltre alle colorazioni specifiche e all’immunoistochimica (identifica la proteina: AL, AA, ATTR…), è utile la biopsia del grasso sottocutaneo, test semplice e poco invasivo per le amiloidosi sistemiche.
🔗 Collegamenti
- Amiloidosi Renale, Amiloidosi Epatica, Amiloidosi Splenica, Amiloidosi Polmonare, Amiloidosi Miocardica — 📋 quadri d’organo
- Amiloidosi AA (Secondaria) — 📋 sottotipo (sistemica reattiva)
- Amiloidosi AL (Primaria) — 📋 sottotipo (sistemica immunocitica)
- Amiloidosi da Dialisi (β₂-microglobulina) — 📋 sottotipo (sistemica)
- Febbre Mediterranea Familiare — 📋 sottotipo (ereditaria)
- Amiloidosi Cerebrale (Alzheimer) — 📋 sottotipo (localizzata cerebrale)
- Proteine di Fase Acuta — 🔗 SAA come precursore dell’amiloide AA
- IL-1, IL-6 — ⬆️ inducono la sintesi epatica di SAA